氨基酚醛树脂基吡咯氮掺杂碳电极材料的制备方法与流程
本发明涉及氨基酚醛树脂基吡咯氮掺杂碳电极材料的制备方法,属于电化学超级电容器技术领域。
背景技术:
超级电容器作为一类新型储能装置,由于其高功率密度和长寿命等特点,受到了业界的广泛关注。而电极材料是影响超级电容器容量的关键因素,其结构和界面特性在增强比容量,循环稳定性,和倍率性能等电容性能方面起着至关重要的作用。通常可以将电极材料分为三类,碳材料,导电聚合物和金属氧化物。其中,碳材料由于其存在形式多样,良好的导电性,质量轻,充放电速度快,稳定性好和廉价易得等优点,成为当前最具前景的电极材料。然而,纯粹碳材料仅能表现出由比表面积决定的双电层电容,其容量依然有待提升(science,2013,341,534-537)。近年来,通过杂原子掺杂,尤其是氮掺杂能够为碳材料提供赝电容活性位点,能够有效提高碳材料的比容量,因此,开发氮掺杂碳材料成为超级电容器领域的研究热点(advancedscience,2017,4,1600408)。
对于碳材料而言,氮原子掺杂的方式一般分为原位掺杂和后掺杂(advancedfunctionalmaterials,2013,23,2322-2328;nanolett,2011,11,2472-2477),其中,原位掺杂方式通常是指采用含氮元素的小分子、聚合物或者生物质材料在碳化过程中将氮原子引入碳骨架中,得到化学结构稳定、掺杂位点均匀、掺杂量可控的氮掺杂碳材料(advancedmaterials,2018,1804394)。氮掺杂形式主要包括吡啶氮、吡咯氮和石墨氮这三种构型,其中,吡啶氮和吡咯氮可以作为赝电容位点提供容量(science,2015,350,1508-1513)。然而,现有技术中,为了提高氮掺杂碳材料导电性,较高的碳化温度常常导致吡咯氮和吡啶氮向石墨氮的不完全转变(electrochimicaacta,2016,205,132-141;journalofmaterialschemistrya,2013,1,6037-6042;acsappl.mater.interfaces,2013,5,2241-2248),导致氮掺杂碳材料中赝电容位点占比下降,从而降低了含氮官能团在超级电容器应用中的利用效率。因此,开发单一吡咯氮、单一吡啶氮或者吡啶氮吡咯氮共掺杂的高容量碳材料成为当前碳电极材料的研究重点和研究难点。为解决这个问题,我们提出了一种氨基酚醛树脂基吡咯氮掺杂碳材料及其制备方法。
技术实现要素:
本发明需要解决的技术问题是提供氨基酚醛树脂基吡咯氮掺杂碳电极材料的制备方法,合成完全具有赝电容活性的单一吡咯氮掺杂碳材料,提高氮掺杂活性位点在赝电容反应中的利用效率,开发具有高比容量和长循环稳定性的氨基酚醛树脂基吡咯氮掺杂碳电极材料。
为解决上述技术问题,本发明所采用的技术方案是:
氨基酚醛树脂基吡咯氮掺杂碳电极材料的制备方法,将3-卤代苯酚,3-氨基苯酚,六亚甲基四胺在常温搅拌,然后水热法聚合得到卤代酚醛树脂,经过水洗干燥,低温碳化,氢氧化钾低温活化,盐酸溶液洗涤、水洗干燥制得。
本发明技术方案的进一步改进在于:包括以下步骤:
a、将3-氨基苯酚、3-卤代苯酚和六亚甲基四胺按一定比例溶解到80ml蒸馏水中形成混合溶液,室温下搅拌一段时间,将混合溶液转移到100ml反应釜中,进行水热反应得到固体样品一,自然冷却至室温后,将固体样品一水洗至ph=7,干燥得到3-氨基苯酚-3-卤代苯酚-甲醛树脂微球样品;
b、将步骤a中所得的3-氨基苯酚-3-卤代苯酚-甲醛树脂微球在氮气气氛中从室温开始升温,热处理一段时间,然后自然冷却至室温收集样品二;
c、将步骤b中所得样品二与活化剂koh按一定比例进行研磨混合均匀,然后在氮气气氛中从室温开始升温,进行活化反应得到固体样品三,自然冷却至室温,将固体样品三用盐酸溶液和水洗涤至ph=7,干燥后得到氨基酚醛树脂基吡咯氮掺杂碳电极材料。
本发明技术方案的进一步改进在于:所述步骤a中3-氨基苯酚与3-卤代苯酚的质量比为1/1~1/7,3-氨基苯酚和3-卤代苯酚的总和与六亚甲基四胺质量比为2/1,混合溶液中六亚甲基四胺的浓度为8.5~9.5×10-3moll-1,室温下搅拌1~1.5小时,其中,3-卤代苯酚为3-氟苯酚、3-氯苯酚或3-溴苯酚。
本发明技术方案的进一步改进在于:所述步骤a混合溶液中六亚甲基四胺的浓度为8.92×10-3moll-1,室温下搅拌1小时。
本发明技术方案的进一步改进在于:所述步骤a中水热反应温度为150~170oc,水热时间为3.5~4.5小时。
本发明技术方案的进一步改进在于:所述步骤a中水热反应温度为160oc,水热时间为4小时。
本发明技术方案的进一步改进在于:所述步骤b中热处理温度为450~520oc,热处理时间为4~5小时。
本发明技术方案的进一步改进在于:所述步骤b中热处理温度为500oc,热处理时间为4小时。
本发明技术方案的进一步改进在于:所述步骤c中样品二与活化剂koh的质量比为1/6,活化反应温度为450~520oc,活化时间为8小时。
本发明技术方案的进一步改进在于:所述步骤c中活化反应温度为500oc。
由于采用了上述技术方案,本发明取得的技术进步是:
本发明在酚醛树脂合成过程中引入氟和氮官能团,低温热处理过程中氟官能团易于脱除,使相邻苯环共价键接,形成具有大共轭结构的碳材料,碱活化过程中碳骨架重排形成单一吡咯氮掺杂碳材料,获得的氨基酚醛树脂基吡咯氮掺杂碳电极材料具有一定导电性,微孔结构和单一吡咯氮掺杂形式,从而表现出高赝电容性能,实现了在低温条件下对氨基酚醛树脂的碳化和活化,保留了大量的单一吡咯氮官能团,提高了氮掺杂的利用率,表现出高容量。
附图说明
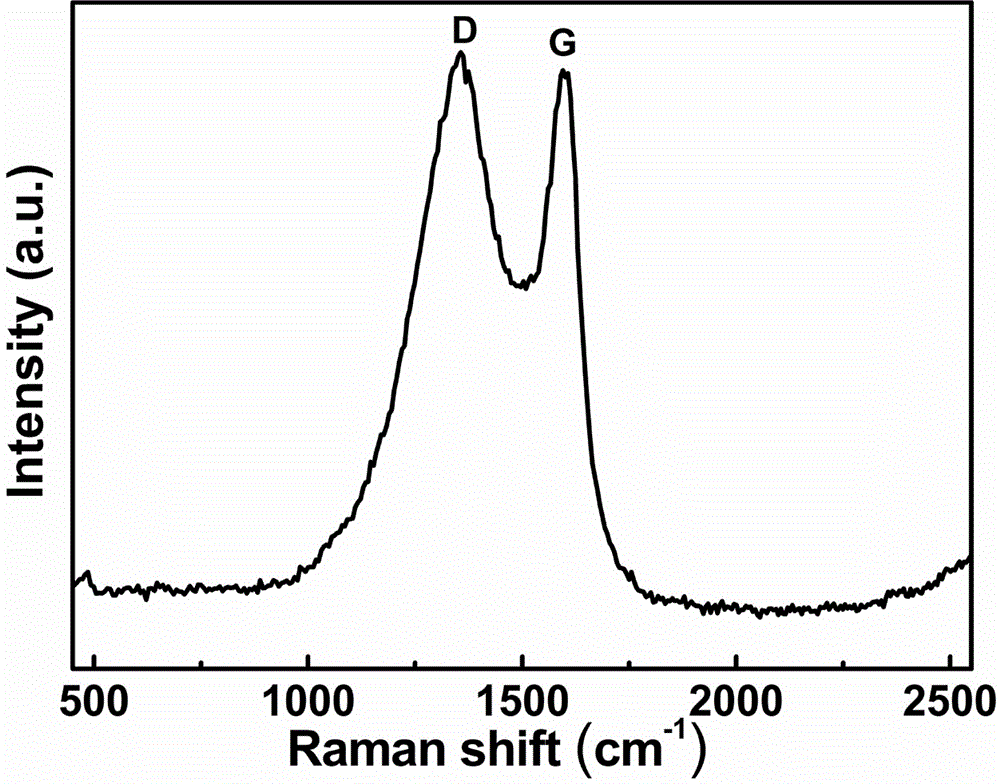
图1是本发明实施例1中氨基酚醛树脂基吡咯氮掺杂碳电极材料的拉曼谱图;
图2是本发明实施例1中氨基酚醛树脂基吡咯氮掺杂碳电极材料的氮气吸脱附曲线和孔径分布谱图;
图3是本发明实施例1中氨基酚醛树脂基吡咯氮掺杂碳电极材料的n1sx射线光电子能谱图(xps);
图4是本发明实施例1中氨基酚醛树脂基吡咯氮掺杂碳电极材料组装的三电极体系在1mol/l硫酸溶液中的恒流充放电曲线;
图5是本发明实施例1中氨基酚醛树脂基吡咯氮掺杂碳电极材料组装的三电极体系在1mol/l硫酸溶液中的循环伏安曲线图;
图6是本发明实施例1中氨基酚醛树脂基吡咯氮掺杂碳电极材料组装的三电极体系在1mol/l硫酸溶液中10a/g电流密度条件下的循环稳定性测试图;
图7是本发明实施例2中氨基酚醛树脂基吡咯氮掺杂碳电极材料的n1sx射线光电子能谱图(xps);
图8是本发明实施例3中氨基酚醛树脂基吡咯氮掺杂碳电极材料的n1sx射线光电子能谱图(xps);
图9是本发明实施例4中氨基酚醛树脂基吡咯氮掺杂碳电极材料的n1sx射线光电子能谱图(xps);
图10是本发明实施例5中氨基酚醛树脂基吡咯氮掺杂碳电极材料的n1sx射线光电子能谱图(xps)。
具体实施方式
下面结合实施例对本发明做进一步详细说明:
氨基酚醛树脂基吡咯氮掺杂碳电极材料的制备方法,将3-卤代苯酚,3-氨基苯酚,六亚甲基四胺在常温搅拌,然后水热法聚合得到卤代酚醛树脂,经过水洗干燥,低温碳化,氢氧化钾低温活化,盐酸溶液洗涤、水洗干燥制得。
具体步骤如下:
a、将3-氨基苯酚、3-卤代苯酚和六亚甲基四胺按一定比例溶解到80ml蒸馏水中形成混合溶液,室温下搅拌一段时间,将混合溶液转移到100ml反应釜中,进行水热反应得到固体样品一,自然冷却至室温后,将固体样品一水洗至ph=7,干燥得到3-氨基苯酚-3-卤代苯酚-甲醛树脂微球样品;
b、将步骤a中所得的3-氨基苯酚-3-卤代苯酚-甲醛树脂微球在氮气气氛中从室温开始升温,热处理一段时间,然后自然冷却至室温收集样品二;
c、将步骤b中所得样品二与活化剂koh按一定比例进行研磨混合均匀,然后在氮气气氛中从室温开始升温,进行活化反应得到固体样品三,自然冷却至室温,将固体样品三用盐酸溶液和水洗涤至ph=7,干燥后得到氨基酚醛树脂基吡咯氮掺杂碳电极材料。
其中,步骤a中3-氨基苯酚与3-卤代苯酚的质量比为1/1~1/7,3-氨基苯酚和3-卤代苯酚的总和与六亚甲基四胺质量比为2/1,混合溶液中六亚甲基四胺的浓度为8.5~9.5×10-3moll-1,优选8.92×10-3moll-1,室温下搅拌1~1.5小时,优选1小时,其中,3-卤代苯酚为3-氟苯酚、3-氯苯酚或3-溴苯酚。水热反应温度为150~170oc,优选160oc,水热时间为3.5~4.5小时,优选4小时。
步骤b中热处理温度为450~520oc,优选500oc,热处理时间为4~5小时,优选4小时。
步骤c中样品二与活化剂koh的质量比为1/6,活化反应温度为450~520oc,优选500oc,活化时间为8小时。
实施例1
a、将0.1g3-氨基苯酚、0.1g3-氟苯酚和0.1g六亚甲基四胺溶解到80ml蒸馏水中,室温下搅拌1h至固体样品完全溶解。然后,将此溶液转移到100ml反应釜中,在160oc下反应4小时,自然冷却至室温后,产物用水洗涤至ph=7,干燥后得到3-氨基苯酚-3-氟苯酚-甲醛树脂微球样品。
b、将步骤a中所得3-氨基苯酚-3-氟苯酚-甲醛树脂微球样品在氮气气氛中从室温加热到500oc,保温4小时。自然冷却至室温后收集样品。
c、将步骤b中所得样品与活化剂koh按质量比1/6进行研磨混合均匀,然后在氮气气氛中,从室温加热到500oc,保温8小时。自然冷却至室温,将固体样品使用盐酸溶液和水洗至ph=7,干燥后得到为氨基酚醛树脂基吡咯氮掺杂碳电极材料。
本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料的拉曼谱图如图1所示,在1353和1593cm-1处各出现一个吸收峰,分别对应碳材料的d带和g带,表明氨基酚醛树脂最终实现了碳化。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料,低温氮气吸附/脱附实验结果如图2所示,其比表面积为450cm2g-1,微孔孔径集中于0.6nm。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料n1sx射线光电子能谱图(xps)如图3所示,表明掺杂氮的构型为单一吡咯态,含量达到4.22at.%。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料,在1mol/l硫酸溶液中的恒流充放电测试结果如图4所示,在电流密度为1a/g时,比电容为618f/g;当电流密度为20a/g时,比电容分别为447f/g,表现出高容量和良好的倍率性能。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料在1mol/l硫酸溶液中进行循环伏安法测试,结果如图5所示,在不同的扫描速率下,循环伏安曲线中存在明显的对称氧化还原峰,具有良好的赝电容行为和电化学可逆性。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料在1mol/l硫酸溶液中进行恒流充放电循环测试,结果如图6所示,在电流密度为10a/g,充放电30000次后,容量保持率为100%,表现出良好的循环稳定性。
实施例2
a、将0.05g3-氨基苯酚、0.15g3-氟苯酚和0.1g六亚甲基四胺溶解到80ml蒸馏水中,室温下搅拌1小时至固体样品完全溶解。然后,将此溶液转移到100ml反应釜中,在160oc下反应4小时,自然冷却至室温后,产物用水洗涤至ph=7,干燥后得到3-氨基苯酚-3-氟苯酚-甲醛树脂微球样品。
b、将步骤a中所得3-氨基苯酚-3-氟苯酚-甲醛树脂微球样品在氮气气氛中从室温加热到500oc,保温4小时。自然冷却至室温后收集样品。
c、将步骤b中所得样品与活化剂koh按质量比1/6进行研磨混合均匀,然后在氮气气氛中,从室温加热到500oc,保温8小时。自然冷却至室温,将固体样品使用盐酸溶液洗涤和水洗至ph=7,干燥后得到为氨基酚醛树脂基吡咯氮掺杂碳电极材料。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料n1sx射线光电子能谱图(xps)如图7所示,表明掺杂氮的构型为单一吡咯态,含量达到3.07at.%。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料,在1mol/l硫酸溶液中进行恒流充放电测试,在电流密度为1a/g时,比电容为472f/g;当电流密度为20a/g时,比电容分别为329f/g,表现出高容量和良好的倍率性能。
实施例3
a、将0.025g3-氨基苯酚、0.175g3-氟苯酚和0.1g六亚甲基四胺溶解到80ml蒸馏水中,室温下搅拌1h至固体样品完全溶解。然后,将此溶液转移到100ml反应釜中,在160oc下反应4小时,自然冷却至室温后,产物用水洗涤至ph=7,干燥后得到3-氨基苯酚-3-氟苯酚-甲醛树脂微球样品。
b、将步骤a中所得3-氨基苯酚-3-氟苯酚-甲醛树脂微球样品在氮气气氛中从室温加热到500oc,保温4小时。自然冷却至室温后收集样品。
c、将步骤b中所得样品与活化剂koh按质量比1/6进行研磨混合均匀,然后在氮气气氛中,从室温加热到500oc,保温8小时。自然冷却至室温,将固体样品使用盐酸溶液洗涤和水洗至ph=7,干燥后得到为氨基酚醛树脂基吡咯氮掺杂碳电极材料。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料n1sx射线光电子能谱图(xps)如图8所示,表明掺杂氮构型为单一吡咯态,含量达到1.51at.%。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料,在1mol/l硫酸溶液中进行恒流充放电测试,在电流密度为1a/g时,比电容为437f/g;当电流密度为20a/g时,比电容分别为388f/g,表现出高容量和良好的倍率性能。
实施例4
a、将0.1g3-氨基苯酚、0.1g3-氯苯酚和0.1g六亚甲基四胺溶解到80ml蒸馏水中,室温下搅拌1小时至固体样品完全溶解。然后,将此溶液转移到100ml反应釜中,在160oc下反应4小时,自然冷却至室温后,产物用水洗涤至ph=7,干燥后得到3-氨基苯酚-3-氯苯酚-甲醛树脂微球样品。
b、将步骤a中所得3-氨基苯酚-3-氯苯酚-甲醛树脂微球样品在氮气气氛中从室温加热到500oc,保温4小时。自然冷却至室温后收集样品。
c、将步骤b中所得样品与活化剂koh按质量比1:6进行研磨混合均匀,然后在氮气气氛中,从室温加热到500oc,保温8小时。自然冷却至室温,将固体样品使用盐酸溶液洗涤和水洗至ph=7,干燥后得到为氨基酚醛树脂基吡咯氮掺杂碳电极材料。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料n1sx射线光电子能谱图(xps)如图9所示,表明掺杂氮构型为单一吡咯态,含量达到2.29at.%。
实施例5
a、将0.1g3-氨基苯酚、0.1g3-溴苯酚和0.1g六亚甲基四胺溶解到80ml蒸馏水中,室温下搅拌1小时至固体样品完全溶解。然后,将此溶液转移到100ml反应釜中,在160oc下反应4小时,自然冷却至室温后,产物用水洗涤直至中性,干燥后得到3-氨基苯酚-3-溴苯酚-甲醛树脂微球样品。
b、将步骤a中所得3-氨基苯酚-3-溴苯酚-甲醛树脂微球样品在氮气气氛中从室温加热到500oc,保温4小时。自然冷却至室温后收集样品。
c、将步骤b中所得样品与活化剂koh按质量比1:6进行研磨混合均匀,然后在氮气气氛中,从室温加热到500oc,保温8小时。自然冷却至室温,将固体样品使用盐酸溶液、水洗和干燥后得到为氨基酚醛树脂基吡咯氮掺杂碳电极材料。
对本实施例中制备的氨基酚醛树脂基吡咯氮掺杂碳电极材料n1sx射线光电子能谱图(xps)如图10所示,表明掺杂氮构型为单一吡咯态,含量达到3.71at.%。
- 还没有人留言评论。精彩留言会获得点赞!