细胞系的制作方法
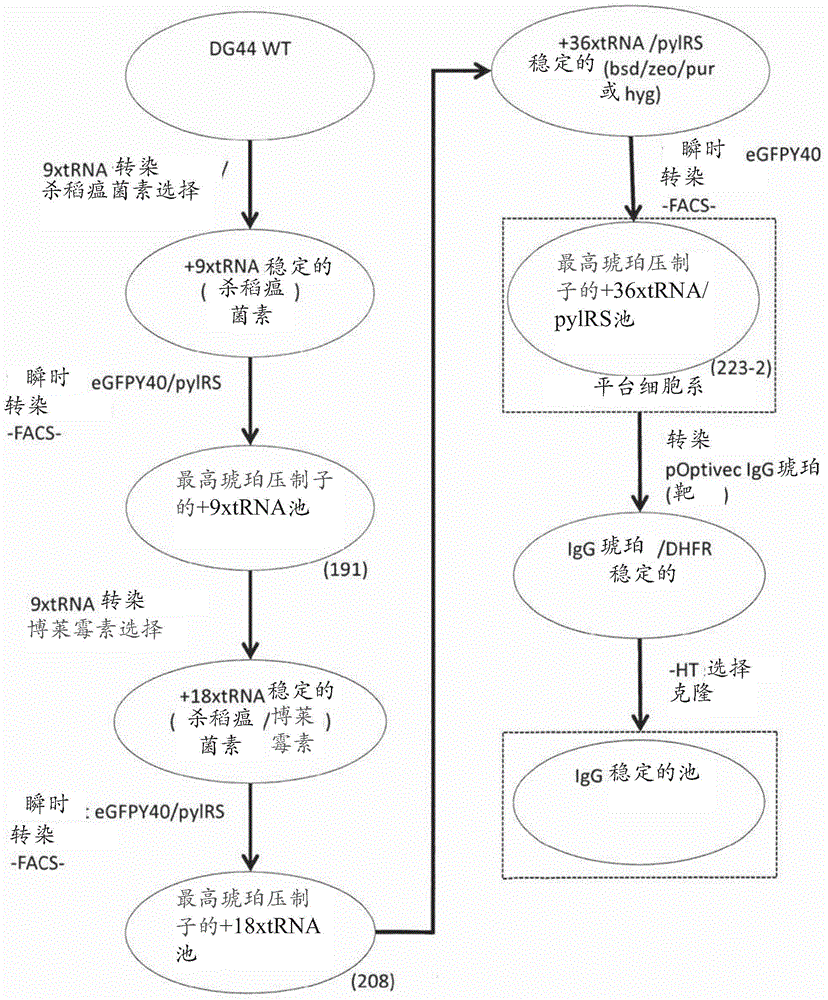
本发明涉及适合用于在将非天然氨基酸掺入蛋白质中使用的稳定的真核细胞系并且涉及制备它们的方法。本发明也涉及具有掺入的非天然氨基酸的蛋白质,该蛋白质适于与其他蛋白质、药物或其他部分共轭,例如以允许半衰期延长并且涉及相应的蛋白质共轭物。进一步地,本发明涉及新颖的氨基酸衍生物。发明背景将非天然氨基酸(nnAA)位点-特异性引入靶蛋白质在产生官能化蛋白质共轭物方面提供了超过非特异性方法的显著优点(王(Wang)等人,2011)。可获得多种包含如下部分的非天然氨基酸,这些部分为共轭化学提供生物正交位点并且使特异性反应能够在这些位点发生。在该共轭位点的位置上进行控制使通过避免活性位点以及必要蛋白质功能结构域而具有最佳功能的产物成为可能。此外,这允许产生均匀并且可预测地修饰的产物,其改善该产物的功能性特征以及纯化。将nnAA位点特异性掺入细菌细胞中已通过氨基酸取代途径以及通过工程化正交氨酰基tRNA合成酶来实现,该正交氨酰基tRNA合成酶仅承担带有非天然氨基酸的其同源tRNA。该非天然氨基酸在靶蛋白质中的位置可通过在该基因序列内的多种密码子指定,但最常见地是针对琥珀密码子。然而,可在大肠杆菌和其他基于原核的系统中表达的多种蛋白质受这些有机体的蛋白质折叠机制限制。真核表达系统(例如哺乳动物表达系统)能够表达更多种类的蛋白质,包括针对最佳治疗功能需要糖基化的那些(例如G-CSF、胰岛素、依泊汀(epoietin)α),作为蛋白质复合体(例如抗体)存在的那些,或需要在细菌系统中不可及的翻译后修饰例如二硫键形成的那些(例如心房利钠因子)。用于将nnAA引入哺乳动物细胞中的系统已通过转染体外承担的tRNA(赫克特(Hecht)等人,1978;戈里尔(Kohrer)等人,2001;戈里尔等人,2003)或使用正交氨酰基tRNA合成酶/tRNA对而遗传编码的tRNA(向井(Mukai)等人2008,刘(Liu)W.等人,2007;王(Wang)W.,2007;耶(Ye),S等人,2008;坂本(Sakamoto),K等人,2002;泷本(Takimoto),J.等人,2009;陈(Chen),P.等人,2009)来开发。化学酰化的tRNA不会被再酰化,并且因此它们被禁止用于无论体外或体内的大规模蛋白质合成。需要基因组编码的RS/tRNA对来正交于该宿主细胞,以保持nnAA插入的特异性。在使用正交氨酰基tRNA合成酶/tRNA对时,该RS和tRNA的正交性是通过在关键位点的突变来实现,以使针对nnAA的特异性成为可能而同时减少或消除对标准氨基酸和宿主tRNA的识别。该tRNA也可被修饰以阻止被宿主RS认知并识别琥珀终止密码子。已开发了若干RS/tRNA对,包括大肠杆菌TyrRS/嗜热脂肪芽孢杆菌tRNAtyr(刘(Liu),W.,2007;耶等人,2008;坂本等人,2002)以及大肠杆菌TyrRS/大肠杆菌tRNAtyr(王(Wang),W.,2007;泷本等人,2009)。已观察到,一个正交RS/tRNA对在针对氨基酸吡咯赖氨酸具有特异性的古细菌(使甲基胺发生分解代谢的产甲烷古细菌)的子集中发生自然进化。吡咯赖氨酸使用了第21氨酰基-tRNA合成酶,天然进化与所有其他氨基酸和tRNA正交。吡咯赖氨酸是天然氨基酸,是真正由琥珀密码子指定的天然氨基酸。布莱特(Blight)等人,2004示出,PylRS和tRNApyl可在大肠杆菌中的琥珀密码子处掺入吡咯赖氨酸。他们还示出,该野生型(“WT”)PyLRS是天然混杂的,并且可掺入赖氨酸的类似物。横山(Yokoyama)等人(EP1911840)证明该PylRS/tRNA系统在真核细胞中是正交的,并且示出若干nnAA掺入由细菌细胞中的琥珀密码子编码的靶蛋白质中。这些作者还鉴别了pylRS中的关键氨基酸残基,这些关键氨基酸残基形成氨基酸结合口袋,并在优于其他标准氨基酸而选择吡咯赖氨酸方面起作用。在此位点的突变产生能够识别并用AzZ-lys氨酰化tRNApy的突变体(柳泽(Yanagisawa)2008)。这种正交性延伸到细菌和真核细胞。PylRS是天然混杂的合成酶,它已经天然进化到排除赖氨酸,但会掺入赖氨酸类似物(不经突变),包括叠氮化物、炔烃和烯烃(柳泽等人,2008;诺伊曼(Neumann)等人,2008;向井(Mukai)等人,2008;阮(Nguyen)等人,2009)。这种特异性的基础依赖于pylRS结合口袋的氨基酸残基与吡咯赖氨酸的吡咯环之间的疏水相互作用,该吡咯赖氨酸的吡咯环稳定并正确地将氨基酸定位在该合成酶的活性位点处(卡佛兰(卡佛兰)等人,2007)。这个RS/tRNA对已通过瞬时转染引入细菌、酵母和哺乳动物细胞中,并显示对于将多个非天然氨基酸掺入到靶蛋白质中是有效的。例如,EP1911840证明将N-ε-boc-赖氨酸掺入到大肠杆菌细胞中的靶蛋白质中。真核细胞的tRNA的表达需要在该tRNA编码序列内的两个内部启动子。这些启动子的共有序列被称为A-盒和B-盒(内伊柯瓦(Naykova)等人,2003)。尽管某些原核衍生的tRNA自然携带作为内部启动子起作用并且可不带有修饰地或带有以下改变地表达于动物细胞中的序列,所述改变是产生基因内启动子序列但不改变该tRNA的功能或它的由其同源RS进行的识别,但是tRNAPyl不包含此类启动子。此外,如通过汉考克(Hancock)等人(2010)在酵母中报导的并且通过诸位发明人在哺乳动物细胞中证实的,其中正常存在A-盒以及B-盒的D环不寻常地小,并且引入所述A-盒以及B-盒会破坏其功能。WO2007099854描述了使用真核snRNA启动子来驱动真核细胞中的tRNAPyl表达。其中所述的DNA构建体包括tRNApyl基因、位于所述tRNA基因的3’的转录终止子序列、以及位于所述tRNApyl基因5’的通过RNA聚合酶II或III诱发转录的启动子序列,例如U1snRNA启动子或U6snRNA启动子。哺乳动物表达是特别感兴趣的,因为它允许产生完全折叠的蛋白质以及蛋白质复合体,像对原核系统或酵母细胞是挑战性的或目前是不可行的全长抗体。在人类(HEK293)和仓鼠(CHO)细胞两者中对编码吡咯赖氨酸氨酰基tRNA合成酶(PylRS)及其tRNApyl的基因进行的瞬时转染实验已显示,在哺乳动物细胞中该PylRS/tRNA对有效地将nnAA掺入靶蛋白质中,在通过琥珀终止密码子指定的位点处(参见例如向井2008)。EP1911840教导了携带WTPylRS的载体、携带tRNApyl基因的载体、以及携带靶基因的载体的引入,其中琥珀突变是在有待插入赖氨酸衍生物的位点处引入的。引入该载体所利用的唯一技术是瞬时转染。实际上,在该专利申请中,未提及对稳定的克隆的选择也未实验式地应用。WO09038195描述了产生突变体PylRS酶以改善其催化活性,并允许掺入具有庞大侧链结构的源自于赖氨酸的非天然氨基酸。具体言之,WO09038195描述了在位置384处的突变(称作马氏甲烷八叠球菌PylRS氨基酸序列),借此Tyr384被苯基丙氨酸(还有其他氨基酸)替代。假设,由于Tyr384与底物氨基酸,特别与其主链相互作用的事实(卡佛兰2007,野泽(Nozawa)2009),存在可独立于该底物增强该酶催化活性的可能性。如上所述,基于PylRS和tRNApyl正交对的表达迄今仅在瞬时稳定的真核细胞系中实现。瞬时稳定的细胞系不适于商业产物的可靠制造;的确,诸位发明人认为,现今市场上的源自于哺乳动物细胞的生物产品是专有地通过稳定细胞系产生的。因此在本领域中仍存在一个需要,来开发用于生产稳定的包含PylRS以及tRNApyl正交对的真核细胞的方法,从而有助于以商业规模生产包含nnAA的蛋白质。本发明着手解决前述需要。吡咯赖氨酸类似物(定义为由天然或遗传进化的PylRS识别并且掺入到琥珀密码子位点处的蛋白质中的氨基酸衍生物)已在过去几年被披露并且已由例如,费克内尔(Feckner)等人(费克内尔(Fekner)、李(Li)&陈(Chan),2010)和刘(Liu)等人综述。已经使用pylRS-tRNApyl系统将具有官能团或翻译后修饰的类似物以位点特异性的方式掺入到蛋白质中。若干研究,参见例如柳泽等人,聚焦于PylRS酶内的突变,以容纳类似物,在这些类似物中N6取代基为该结合口袋吡咯赖氨酸内的芳族环。其他人,例如阮等人(也于WO2010/139948中)和李(Li)等人(也于WO2011/044255中)聚焦于不携带庞大N6取代基的吡咯赖氨酸类似物的鉴别,结果获得更简单的类似物,这些更简单的类似物对于合成天然pylRS/tRNApyl对和与之相互作用是简单的。仍存在开发另外的吡咯赖氨酸类似物的需要。在迄今为止制造的吡咯赖氨酸类似物被局限于从赖氨酸骨架发展的那些的时候,诸位发明人已起始于各种氨基酸结构产生了成功掺入到具有天然pylRS/tRNApyl对的蛋白质中的吡咯赖氨酸类似物。在最近几年中已经开发了由通过合成连接体共价结合至高细胞毒性药物的重组嵌合人源化或人抗体构成的抗体药物共轭物(ADC),以将细胞毒性药物靶向肿瘤细胞。靶向肿瘤关联抗原的抗体、有效力的毒素以及适当的共轭化学的正确组合可在直接将该毒素递送到这些肿瘤细胞方面非常有效,同时避免了该药物对正常组织的毒性。发展至今的ADC是共轭以及非共轭抗体的异质混合物,取决于当产生该ADC时所使用的共轭化学。具体言之,最常用的共轭方案的随机性导致具有每抗体分子(DAR)共轭变化数目的药物连同变化的共轭位点的种类的集合。常用的共轭化学包括基于赖氨酸侧链的共轭,其导致大范围的种类,这归因于赖氨酸残基在典型抗体中的大的可用性。已通过工程化半胱氨酸残基以产生反应性硫醇基团而获得更位点特异性的共轭,得到近乎均匀的ADC。Her2肿瘤关联抗原,即EGFR家族的一员,已在乳腺癌中用赫塞汀(一种抗-Her2抗体)成功靶定,然而,该抗体本身在有限群组的患者中有效。现在可获得更高效的形式,即阿恩曲珠单抗(ado-trastuzumabemtansine),具有关联至其的毒素。阿恩曲珠单抗能够有效地治疗赫塞汀难以治疗的患者,归因于阿恩曲珠单抗将毒素递送到癌细胞的细胞质中的能力。由阿恩曲珠单抗和布吐西单抗维多汀(Brentuximabvedotin)使用的共轭化学利用了已有的通常形成二硫键的半胱氨酸残基以及新近工程化的游离半胱氨酸残基。这种途径已经导致产生在mAb上不同的位置处具有不同数目药物的ADC的异质混合物。所使用的连接体包括硫醚(Kadcyla)连同二肽连接体(Adcetris),后者被溶酶体酸水解酶特异性地裂解。两种类型的连接体似乎是有效的,但不是最佳的。常规的Cys或Lys定向的生物共轭方法,例如用于通过阿恩曲珠单抗、布吐西单抗维多汀、以及吉妥珠单抗奥佐米星进行制造的那些方法,允许在遭遇肿瘤细胞之前提前释放毒素。在2000年批准的吉妥珠单抗奥佐米星在2010年从市场撤回,归因于使用了酸不稳定的连接体的高毒性,该酸不稳定的连接体引起在血液中毒素从ADC中不可忍受的释放。因此,在本领域中仍存在需要来开发高度均匀的ADC,其中共轭位点以及每抗体的药物数目受到良好控制。诸位发明人已经观察到,通过使用nnAA的位点特异性掺入以及随后的抗体在nnAA的位点的共轭,可能产生均匀且高效的ADC。此外,诸位发明人已经发现了在IgG恒定区内的如下位点,这些位点可被用于共轭而不破坏该抗体的结合特异性或其体内药代动力学性质。发明概述根据本发明,提供了用于制备稳定的真核细胞系的方法,该真核细胞系表达pylRS以及tRNApyl并且适于掺入许可表达靶蛋白质的基因,该靶蛋白质包含一个或多个由琥珀密码子编码的非天然氨基酸。本发明是源自于诸位发明人的以下发现,这些发现涉及通过现有技术方法制备的细胞系的不稳定性的来源。可观察到,常规的真核细胞,例如表达pylRS以及tRNApyl而不具有任何许可表达包含一个或多个由琥珀密码子编码的非天然氨基酸的靶蛋白质的基因的CHO以及HEK293细胞采用指示细胞毒性的表型,包括培养物中更高比例的死细胞、圆形的细胞形态、细胞与生长板的宽松附着、以及降低的细胞生长速率。诸位发明人观察到,在随后表达所述许可表达靶蛋白质的基因后,细胞的健康状况似乎改善明显。诸位发明人看到,该毒性与高量的tRNApyl在该系统中的表达相关,在不存在nnAA的情况下这诱导在琥珀终止密码子中终止的必需管家基因的延伸(利伯曼(Liebman)以及谢尔曼(Sherman)1976;利伯曼等人,1976)。因此,不受理论的限制,tRNApyl的毒性作用可以是当不存在靶蛋白质的情况下存在高水平的tRNApyl时发生的不完全正交性的结果。虽然该tRNApyl在哺乳动物细胞中大多数情况下正交,可能的是,在不存在nnAA的情况下,该tRNApyl可被宿主RS中的一个无效率地氨酰基化(此时天然酶,pylRS是空缺的)。在表达高水平的tRNA的细胞中,可产生显著量的氨酰基化的tRNA,其强加必需基因的不规则琥珀压制。因而,诸位发明人推测,稳定细胞系可通过以减少或掩蔽了tRNApyl的明显毒性作用作为其目标的方法来产生。因此,作为本发明的第一个方面,提供了用于使表达PylRS和tRNAPyl并且适于掺入编码靶蛋白质的基因的真核细胞系(特别是哺乳动物细胞系)稳定化的方法,该靶蛋白质包含一个或多个由琥珀密码子编码的非天然氨基酸,该方法包括在tRNAPyl表达对细胞生活力和/或细胞生长的不利影响被减小或被消除的条件下培养所述细胞系。作为本发明的第二个方面,提供了如此处所述的具有式VII的诱饵氨基酸(dnnAA),当在产生或维持所述稳定细胞系期间添加到培养基中时其具有减少或掩蔽tRNApyl的毒素作用的优点。根据本发明的第三个方面,提供了如此处所述的具有式V以及VI的吡咯赖氨酸的新颖的非天然氨基酸类似物,其具有制备起来简单明了、容易掺入到蛋白质中(典型地当适当使用时不损失生物活性)并且提供用于生物共轭的有用手段的优点。根据本发明的第四个方面,提供了通过在预先确定的位置位点特异性地插入非天然氨基酸来获得高度均匀以及具有活性的抗体药物共轭物的方法。附图简要说明图1.方案,详述了针对开发平台细胞系所进行的反复引入RS/tRNA元件和选择步骤以及随后的表达细胞系。图2.亲本以及分选细胞的功能比较,这些细胞是在开发平台细胞系时分离的。图3.从稳定细胞系表达的、包含lys-叠氮化物nnAA的抗-IL-6IgGK274的表征。图4.对tRNA如何成为该系统的限制组分以及背景活性如何具有抑制细胞生长作用的说明。图5.非还原SDS-PAGE胶,线性20kPEG炔烃与抗-IL-6AzAb的PEG化(A)以及非还原SDS-PAGE胶,20KPEG环辛炔与抗-IL-6-Lys叠氮化物274h的PEG化(B)。图6.非还原SDS-PAGE胶,抗-IL-6-Lys叠氮化物274h至31A12-20KPEG炔烃的双特异性地共轭(A),以及还原SDS-PAGE胶,抗-IL-6-Lys叠氮化物274h至31A12-20KPEG炔烃的双特异性地共轭(B)。图7.在位置R131处包含炔丙基-赖氨酸nnAA的FGF21被有效地PEG化并且保持体内功能的证据。图8.毒素共轭的抗体和抗体片段显示体外特异性活性的证据。图9.非还原胶,包含叠氮化物的抗-Her2抗体与20KPEG环-炔烃发生反应(A),以及还原胶,包含叠氮化物的抗-Her2抗体与20KPEG环-炔烃发生反应(B)。图10.非还原SDS-PAGE胶,抗-Her2-Lys叠氮化物274h70l的20KPEG化(A)以及还原SDS-PAGE胶,4D5AzAb-4(B)的20KPEG化。图11.还原胶,包含叠氮化物的抗-Her2抗体在存在铜的情况下与20KPEG末端炔烃发生反应(A),以及非还原胶,包含叠氮化物的V抗-Her2抗体在存在铜的情况下与20KPEG末端炔烃发生反应(B)。图12.还原胶,掺入了Lys-叠氮化物、共轭至20K线性PEG环辛炔的抗-PSMAscFv(A),以及还原胶,具有nnAALys-叠氮化物、共轭至MMAF-VCP-环辛炔的抗-PSMAscFv(B)。图13.体外功能性测定检查与lys叠氮化物竞争的诱饵nnAA的功能以及特异性。图14.体外功能性测定检查与lys叠氮化物竞争中dnnAA功能的效力以及其对包含pylRS/tRNA的细胞中的背景琥珀压制的影响。图15.在存在或不存在诱饵nnAA的情况下生长的包含pylRS/tRNA的细胞的生长速率以及生活力测定。图16.用诱饵nnAA处理的细胞中pylRS/tRNA功能的群体分析。图17.在用诱饵nnAA处理的培养物中检查包含pylRS/tRNA对的细胞的生长速率。图18.包含单克隆抗体的叠氮化物的PEG化。泳道1:未经处理的抗体,泳道2:具有掺入到重链中的吡咯赖氨酸类似物式V.1并经受PEG化条件的抗体;泳道3:具有掺入到重链中的吡咯赖氨酸类似物式VI.1并经受PEG化条件的抗体。图19.具有本来包含掺入到重链中的吡咯赖氨酸类似物式VI.1的抗体的4D5-阿里他汀F抗体药物共轭物的HIC色谱图。图20.PEG化4D5位置性突变体的SDSPAGE分析图21.通过HIC色谱法进行的阿里他汀(Auristatin)与在不同的位置掺入了叠氮化物的4D5-AzAb的共轭的反应分析。图22.源自于4D5叠氮化物的位置性突变体的ADC的SDS-PAGE图23.通过体外细胞毒测定进行的4D5-2AzAb阿里他汀抗体药物共轭物的位置变体对高和低表达Her2细胞系的效价以及选择性的评估。图24.通过HIC色谱法进行的4D5-2AzAb(HC274)与荧光染料的共轭的反应分析。图25.在位置H274修饰的4D5的药代动力学和稳定性图26.非共轭抗体以及用阿里他汀环辛炔衍生物制备的4D5-2AzAb(HC274)-阿里他汀F抗体药物共轭物的HIC色谱图的叠加。图27.4D5-2AzAb(HC274)-AF共轭物针对Her2阳性肿瘤细胞系的体外抗肿瘤活性。图28.4D5-2AzAb(HC274)-AF的体内抗肿瘤活性。表达为针对每个组的平均肿瘤尺寸(A)的或存活百分比(B)的肿瘤进展。图29.4D5-2AzAb/FGF21双特异性物的SDSPAGE分析。泳道1:MW标记,泳道2:未经处理的FGF21,泳道3:4D5-FGF21双特异性反应,泳道4:4D5-2AzAb。图30.针对用在位置H274(抗-Her2)处包含nnAA的全长mAb以及针对IL6的scFv构建的双特异性抗体的检测的ELISA测定方案和数据。A)ELISA显示捕获该全长mAb(4D5)并且使用IL6检测该双特异性物。B)ELISA显示该全长mAb(4D5)功能性结合至Her2的细胞外结构域,并且随后检测该mAb。C)ELISA测定显示该mAb功能性结合至该Her2细胞外结构域并且scFv结合至IL6。图31.4D5AzAb-FGF21双特异性物的SDSPAGE分析图32.在CuAAC条件以及TBTA下,4D5-2AzAb(HC274)的20kDaPEG化的反应混合物的SDS-PAGE分析图33.用CuAAC以及THPTA进行的20kPEG化至4D5AzAb的产物的SDSPAGE分析图34.A,B,在还原和非还原条件下,用CuAAC/THPTA进行的4D5-2AzAb(HC274)的2kDaPEG化的PAGE分析。C,4D5-AzAb的2kDaPEG化的最终反应混合物的HIC色谱图图35.DAR44D5-AFADC的体外细胞毒性影响图36.4D5-鹅膏蕈碱ADC的体外细胞毒性影响图37.通过CUAAC以及SPAAC化学获得的4D5-AFADC的体外细胞毒性影响。图38.在CuAAC条件下,4D5-2AzAb(HC274)共轭至阿里他汀F衍生物的反应混合物的HIC色谱图。图39.用20KDaPEG炔烃位点特异性地修饰的赫塞汀AzAb重链的凝胶迁移率测定。图40.共轭至阿里他汀F-环辛炔的赫塞汀AzAb的分析。A)未经处理的赫塞汀-AzAb和共轭反应的产物的HIC分析。B)在还原(B)和非还原(C)条件下,通过SDS-PAGE进行的未经处理下的以及共轭反应下的凝胶迁移率测定。图41.共轭至阿里他汀F-炔烃的赫塞汀AzAb的分析。A)未经处理的赫塞汀-AzAb和共轭反应的产物的HIC分析。B)在还原(B)和非还原(C)条件下,通过SDS-PAGE进行的未经处理下的以及共轭反应下的凝胶迁移率测定。图42.赫塞汀ADC的体外细胞毒性序列表的简要说明SEQIDNo1:PylRS马氏甲烷八叠球菌WT氨基酸序列SEQIDNo2:PylRS马氏甲烷八叠球菌,Y384F突变体氨基酸序列SEQIDNo3:PylRS巴氏甲烷八叠球菌WT氨基酸序列SEQIDNo4:PylRS哈夫尼脱亚硫酸菌(Desulfitobacteriumhafniense)氨基酸序列SEQIDNo5:PylRS埃斯蒂瓦兰斯甲烷八叠球菌(Methanosarcinaacetivorans)氨基酸序列SEQIDNo6:PylRS布氏拟甲烷球菌(Methanococcoidesburtonii)氨基酸序列SEQIDNo7:PylRS嗜热甲烷八叠球菌(Methanosarcinathermophila)氨基酸序列SEQIDNo8:PylRS织里甲烷咸菌(Methanosalsumzhilinae)氨基酸序列SEQIDNo9:PylRS伊夫氏甲烷盐菌(Methanohalobiumevestigatum)氨基酸序列SEQIDNo10:PylRS马希甲烷嗜盐菌(Methanohalophilusmahii)氨基酸序列SEQIDNo11:PylRS吉布森脱硫肠状菌(Desulfotomaculumgibsoniae)氨基酸序列SEQIDNo12:PylRS梅里迪维脱硫芽孢弯曲菌(Desulfosporosinusmeridiei)氨基酸序列SEQIDNo13:PylRS乙酸氧化脱硫肠状菌(Desulfotomaculumacetoxidans)氨基酸序列SEQIDNo14:PylRS马氏甲烷八叠球菌WT核苷酸序列SEQIDNo15:PylRS马氏甲烷八叠球菌,Y384F突变体核苷酸序列SEQIDNo16:PylRS密码子优化的马氏甲烷八叠球菌核苷酸序列SEQIDNo17:PylRS巴氏甲烷八叠球菌核苷酸序列SEQIDNo18:PylRS哈夫尼脱亚硫酸菌(Desulfitobacteriumhafniense)核苷酸序列SEQIDNo19:PylRS埃斯蒂瓦兰斯甲烷八叠球菌(Methanosarcinaacetivorans)核苷酸序列SEQIDNo20:PylRS布氏拟甲烷球菌(Methanococcoidesburtonii)核苷酸序列SEQIDNo21:PylRS嗜热甲烷八叠球菌核苷酸序列SEQIDNo22:PylRS织里甲烷咸菌(Methanosalumzhilinae)核苷酸序列SEQIDNo23:PylRS伊夫氏甲烷盐菌(Methanohalobiumevestigatum)核苷酸序列SEQIDNo24:PylRS吉布森脱硫肠状菌(Desulfotomaculumgibsoniae)核苷酸序列SEQIDNo25:PylRS马希甲烷嗜盐菌(Methanohalophilusmahii)核苷酸序列SEQIDNo26:tRNApyl巴氏甲烷八叠球菌SEQIDNo27:tRNApyl埃斯蒂瓦兰斯甲烷八叠球菌(Methanosarcinaacetivorans)SEQIDNo28:tRNApyl马氏甲烷八叠球菌SEQIDNo29:tRNApyl布氏拟甲烷球菌(Methanococcoidesburtonii)SEQIDNo30:tRNApyl哈夫尼脱亚硫酸菌(Desulfobacteriumhafniense)SEQIDNo31:H1/TO启动子SEQIDNo32:U6snRNA启动子SEQIDNo33:SNR52启动子SEQIDNo34:H1启动子SEQIDNo35:U6-tRNApyl构建体SEQIDNo36:GFP核苷酸序列SEQIDNo37:GFP氨基酸序列SEQIDNo38:GFPY40氨基酸序列SEQIDNo39:抗-IL-6(28D2)γ核苷酸序列SEQIDNo40:抗-IL-6(28D2)γ氨基酸序列SEQIDNo41:抗-IL-6(28D2)γ_琥珀K274核苷酸序列SEQIDNo42:抗-IL-6(28D2)γ_琥珀K274氨基酸序列SEQIDNo43:抗-IL-6(28D2)κ核苷酸序列SEQIDNo44:抗-IL-6(28D2)κ氨基酸序列SEQIDNo45:抗-Her2(4D5)γ核苷酸序列SEQIDNo46:抗-Her2(4D5)γ氨基酸序列SEQIDNo47:抗-Her2(4D5)γ_K274琥珀核苷酸序列SEQIDNo48:抗-Her2(4D5)γ_K274琥珀氨基酸序列SEQIDNo49:抗-Her2(4D5)γ_T359琥珀核苷酸序列SEQIDNo50:抗-Her2(4D5)γ_T359琥珀氨基酸序列SEQIDNo51:抗-Her2(4D5)κ核苷酸序列SEQIDNo52:抗-Her2(4D5)κ氨基酸序列SEQIDNo53:抗-Her2(4D5)κD70琥珀核苷酸序列SEQIDNo54:抗-Her2(4D5)κD70琥珀氨基酸序列SEQIDNo55:抗-Her2(4D5)κE81琥珀核苷酸序列SEQIDNo56:抗-Her2(4D5)κE81琥珀氨基酸序列SEQIDNo57:抗-PSMAscFv核苷酸序列SEQIDNo58:抗-PSMAscFv氨基酸序列SEQIDNo59:抗-PSMAscFv_117琥珀核苷酸序列SEQIDNo60:抗-PSMAscFv_117琥珀氨基酸序列SEQIDNo61:FGF21WT核苷酸序列SEQIDNo62:FGF21WT氨基酸序列SEQIDNo63:FGF21R131琥珀核苷酸序列SEQIDNo64:FGF21R131琥珀氨基酸序列SEQIDNo65:FGF21F12琥珀核苷酸序列SEQIDNo66:FGF21F12琥珀氨基酸序列SEQIDNo67:FGF21L60琥珀核苷酸序列SEQIDNo68:FGF21L60琥珀氨基酸序列SEQIDNo69:FGF21P90琥珀核苷酸序列SEQIDNo70:FGF21P90琥珀氨基酸序列SEQIDNo71:FGF21P140琥珀核苷酸序列SEQIDNo72:FGF21P140琥珀氨基酸序列SEQIDNo73:GFPY40核苷酸序列SEQIDNo74:赫塞汀核苷酸序列重链SEQIDNo75:赫塞汀氨基酸序列重链SEQIDNo76:赫塞汀H274核苷酸序列重链SEQIDNo77:赫塞汀H274氨基酸序列重链SEQIDNo78:赫塞汀核苷酸序列轻链SEQIDNo79:赫塞汀氨基酸序列轻链SEQIDNo80:3xFlag标签序列SEQIDNo81:5xPro-6xHis标签SEQIDNo82:人IgG1重链的部分(恒定区)SEQIDNo83:SEQIDNo52的部分(框架区)SEQIDNo84:SEQIDNo52的部分(框架区)发明详述定义术语“烷基”是指脂肪族键或取代基,典型地包含1-6个例如1-4个碳原子,并且可以是直链或支链的。实例包括甲基、乙基、正丙基、异丙基、正丁基和叔丁基。术语“烯基”是指脂肪族键或取代基,典型地包含2-6个例如2-4个碳原子,并且可以是直链或支链的,并且就包含至少一个C=C部分而言是不饱的。特异性实例是末端烯烃基团,其中C=C部分在该末端。烯基的实例包括乙烯基、丙烯-1-基、丙烯-2-基、以及2-甲基-丙烯-2-基。术语“炔烃”或“炔基”是指脂肪族键或取代基,典型地包含2-6个例如2-4个碳原子,并且可以是直链或支链的,并且就包含至少一个C≡C部分而言是不饱的。具体实例是末端炔烃基团,其中C≡C部分在该末端。炔基基团的实例包括-C≡CH和-C≡C-CH3。术语“芳基”是指可以是键的部分或取代基的部分的芳族环结构。芳基部分可包含一个环(例如苯基)或两个环(例如萘基)或多于两个环,条件是至少一个环是芳族。芳基基团可以被例如一个或多个(例如一个或两个,例如一个)取代基取代,该取代基选自烷基、烯基、炔基、氟烷基、卤素、烷氧基、硝基和氰基。示例性的芳基是苯基。术语“杂芳基”是指可以是键的部分或取代基的部分的杂芳族环结构。该杂芳族环可包含1-4个(更通常1-3个例如一个或两个)选自O、N和S的杂原子。杂芳基部分可包含一个环或两个环或多于两个环,条件是至少一个环是杂芳族。包含一个6元环的示例基团包括吡啶和嘧啶。包含一个5元环的示例基团包括吡咯、呋喃、噻吩、噁唑、噻唑、二唑、噻二唑和四唑。包含两个环的杂芳基部分可在一个或两个环中包含杂原子。实例包括喹啉和异喹啉。杂芳基基团可以被例如一个或多个(例如一个或两个,例如一个)取代基取代,该取代基选自烷基、烯基、炔基、氟烷基、卤素、烷氧基、硝基和氰基。术语“甲基”或“Me”是指CH3基团术语“OMe”是指O-CH3基团。术语“乙基”或“Et”是指CH2CH3基团。术语“OEt”是指O-CH2CH3基团术语“tBu”是指C(CH3)3基团术语“OtBu”是指O-C(CH3)3基团。术语“OBn”或“O苄基”是指O-CH2-Ph基团术语“OFmoc”或“OCH2芴”是指以下结构:术语“苯基”或“Ph”是指如以下结构中的苯环:术语“烯丙基”是指CH2-CH=CH2基团术语“乙基氯化物”是指CH2CH2-Cl基团术语“叠氮化物”和“叠氮基”是指N=N(+)=N(-)或N3官能团。术语“叠氮基烷基”意指被叠氮基、尤其末端叠氮基取代的烷基。实例包括–(CH2)nN3,其中n=1-4。术语“卤代烷基”意指被一个或多个(例如1、2或3个,尤其1或2个例如1个)卤素原子(例如Cl或F原子)取代的烷基。实例包括-CF3以及-CH2CH2Cl。术语“炔丙基”是指附属到末端炔烃的甲基基团。它由-CH2-C≡C-H表示。术语“酰胺”是指–C(=O)-NH-键。术语“氨基甲酸酯”是指–O-C(=O)-NH-键。术语“酯”是指–C-C(=O)-O-C键术语“烷氧基”是指基团–O-烷基。术语“酮”是指C-C(=O)-C键。术语“吡咯赖氨酸类似物”意指由天然或遗传进化的PylRS识别并且掺入到蛋白质中的无义密码子位点处的氨基酸衍生物。术语“这20个天然氨基酸中一个的侧链”是指涉及由其单个字母代码A、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、W和Y所知道的20个天然氨基酸的式HOOC-CHR-NH2中的基团R。预期了L或S立体化学(或其混合物),但是优选L立体化学。术语“细胞生活力”是指在体外培养的细胞的背景下,基于整体细胞样本确定活(有生活力的)或死细胞。如果具有生长以及发育的能力,则细胞被认为是有生活力的。生活力测定是基于有生活力的细胞的物理特性例如膜完整性,或其代谢活性。术语“细胞生长”是指如通过经一段时间细胞分裂的数目测量的细胞增殖。生长是通过追踪随着时间的推移培养物的细胞密度(细胞/mL)测量的。术语“稳定表达”是指通过将感兴趣的基因(或相应的cDNA)整合到靶细胞的染色体中实现的蛋白质的表达。因此术语“稳定的整合”是指将感兴趣的基因(或相应的cDNA)整合到靶细胞的染色体中:最初,必须将感兴趣的基因引入该细胞,随后引入核中,并且最终必须把它整合到染色体DNA中。可选择并且以不同方式培养稳定转染的细胞:例如,选择性标记是在相同的或在第二个经共转染的载体上共表达的。存在多个用于选择经转染的细胞的系统,包括对抗生素例如新霉素磷酸转移酶的抗性,赋予对G418、二氢叶酸还原酶(DHFR)、或谷氨酰胺合成酶的抗性。培养经转染的细胞可以大批进行以获得抗性细胞的混合群体,或通过单一细胞培养进行以从单一整合事件获得细胞克隆。术语“靶基因”是指编码通过插入nnAA修饰的蛋白质的基因。根据本发明的不同实施例“诱饵氨基酸”途径根据实施例,提供了一种方法,其中tRNApyl对细胞生活力和/或细胞生长的不利影响被减小或消除的条件包括如下条件,其中该细胞系所培养的培养基中存在有诱饵氨基酸,该诱饵氨基酸是PylRS的底物(即是氨酰基化的并且装载到该tRNApyl上),但不能掺入延伸的蛋白质链中。因此,将非天然氨基酸掺入蛋白质的方法可包括以下步骤:1.将诱饵氨基酸引入容易被针对该同源tRNA的正交RS识别并且活化的细胞的生长培养基中2.将该RS以及tRNA引入真核细胞,在一个或多个质粒上3.针对包含该RS以及tRNA表达盒的细胞进行选择4.分离一种或多种表达该RS蛋白质以及tRNA的稳定克隆,从而产生平台细胞系。选择能够有大于30%,例如大于40%或50%或60%或70%或80%或优选地大于90%的非天然氨基酸掺入率的细胞。5.引入靶蛋白质的cDNA,借此已在待掺入该非天然氨基酸的一个或多个位置处引入一个或多个琥珀密码子6.分离表达高水平(大于0.5-10pg/细胞/天)的靶基因产物的稳定的克隆7.在不存在该诱饵氨基酸但存在允许在该琥珀密码子处掺入的非天然氨基酸的情况下,使该细胞系生长。根据此实施例,当该诱饵氨基酸存在于包含PylRS以及tRNA合成酶对的细胞系中时,它结合至PylRS并且氨酰基化至tRNA。然后其传递到核糖体上,此处tRNA结合琥珀密码子,但使得酰化的氨基阻滞的诱饵不能形成肽键,因此该蛋白质在此位点终止。在一个实施例中,一旦靶蛋白质的cDNA被引入细胞中诱饵就不再存在。可替代地,当编码靶蛋白质的cDNA被引入该细胞中时,诱饵氨基酸被维持在培养基中。在可替代的实施例中,靶蛋白质的表达发生在存在诱饵氨基酸的情况下。因此,根据此实施例,在靶蛋白质产生时,将诱饵氨基酸以及PylRS优先使用的所希望的非天然氨基酸两者都添加到(或存在于)发酵培养基中。如果诱饵氨基酸与所希望的非天然氨基酸竞争结合到PylRS至任何显著程度的话,它不应被添加到或存在于发酵培养基中。在另一个实施例中,靶蛋白质的表达在存在诱饵氨基酸的情况下不会发生(例如在从发酵培养基消除之后)。因此,根据此实施例,在靶蛋白质表达期间,诱饵氨基酸不被引入发酵培养基中。在靶蛋白质产生期间,仅将优先结合PylRS的所希望的非天然氨基酸添加到(或存在于)发酵中。根据此实施例,贯穿对包含PylRS/tRNA的平台细胞系的选择以及分离,在培养基中利用诱饵氨基酸。引入并选择包含一个或多个琥珀密码子的靶基因之后,将诱饵氨基酸去除。如果希望的话,可以采用多个(例如2、3、4、5、6或7个或更多个)诱饵氨基酸。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系能够表达PylRS以及tRNAPyl,并且该真核细胞系适于掺入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,该方法包括:(a)在一个或多个步骤中向真核细胞系中引入编码PylRS以及tRNAPyl的基因,并且使得PylRS以及tRNAPyl稳定表达于所述细胞系;(b)在存在诱饵氨基酸的情况下培养或选择所得的细胞系,该诱饵氨基酸是PylRS的底物,但不能够掺入延伸的蛋白质链中,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系能够表达PylRS、tRNAPyl以及包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质,该方法包括:(a)在一个或多个步骤中向真核细胞系中引入编码PylRS以及tRNAPyl的基因,以使得PylRS以及tRNAPyl稳定表达于所述细胞系;(b)在存在诱饵氨基酸的情况下培养或选择所得的细胞系,该诱饵氨基酸是PylRS的底物,但不能够掺入延伸的蛋白质链中,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响;(c)将编码包含一个或多个非天然氨基酸的靶蛋白质的基因引入所述真核细胞系,以使得所述靶蛋白质稳定表达于所述细胞系中;并且(d)在不存在所述诱饵氨基酸的情况下表达该靶蛋白质。本发明的另一个方面是用于产生根据前述方面中任一个所述的稳定的真核细胞系的方法,其中对该细胞系进行的培养或选择是在存在诱饵氨基酸的情况下进行,该诱饵氨基酸是pylRS的底物,但不能够掺入延伸的蛋白质链中,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响。“靶标第一”途径根据可替代的实施例,提供了如下方法,其中tRNAPyl对细胞生活力和/或细胞生长的不利影响被减小或被消除的条件包括如下条件,其中也通过所述细胞系表达包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质。因此,将非天然氨基酸掺入蛋白质的方法可包括以下步骤:1.在一个或多个质粒上将包含琥珀密码子的靶基因在非天然氨基酸有待被掺入的位置引入到真核细胞中2.分离以高水平(大于0.5或大于10pg/细胞/天)表达靶蛋白质的细胞池或克隆池3.在一个或多个质粒上将RS以及tRNA引入这些细胞,并且针对包含RS以及tRNA的克隆进行选择4.在存在允许在琥珀密码子处掺入的非天然氨基酸的情况下使该细胞系生长,并且分离显示非天然氨基酸在该所希望的位点的掺入效率大于30%,例如大于40%或50%或60%或70%或80%或优选地大于90%的细胞。另外的实施例是稳定的真核细胞系,该真核细胞系表达PylRS以及tRNAPyl,并且还在诱导型启动子控制下表达包含一个或多个由琥珀密码子编码的非天然氨基酸的诱饵蛋白质。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系表达PylRS、tRNAPyl以及包含一个或多个由琥珀密码子编码的非天然氨基酸的靶蛋白质,该方法包括:(a)将编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因引入真核细胞系,以使得所述基因稳定整合于所述细胞系中;(b)在一个或多个步骤中另外的将编码PylRS以及tRNAPyl的基因引入所述细胞系,以使得PylRS以及tRNAPyl稳定表达于所述细胞系中;并且(c)在tRNAPyl仅由所述细胞系表达同时该靶蛋白质也由所述细胞系表达的条件下,在存在一个或多个非天然氨基酸的来源的情况下,培养所得的细胞系,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响。“阻抑型tRNA”途径根据可替代的实施例,提供了如下方法,其中,这些其中tRNAPyl对细胞生活力和/或细胞生长的不利影响被减小或被消除的条件包括如下条件,其中tRNAPyl的表达在阻抑型启动子控制下发生。还提供了真核细胞系,该真核细胞系表达或能够表达PylRS以及tRNAPyl,其中tRNAPyl的表达在阻抑型启动子控制下发生。因此,将非天然氨基酸掺入蛋白质的方法可包括以下步骤:1.在一个或多个质粒上将RS以及tRNA引入真核细胞中,后者包含使能够控制tRNA表达的阻抑型启动子元件。2.在阻抑条件下针对包含RS以及tRNA表达盒的细胞进行选择3.诱导tRNA表达,并且分离一个或多个表达高水平的RS蛋白质和tRNA或证明有效压制报告基因中的琥珀密码子的稳定的克隆,从而产生平台细胞系。选择能够有大于30%,例如大于40%或50%或60%或70%或80%或优选地大于90%的非天然氨基酸掺入率的细胞。4.引入靶蛋白质的cDNA,借此已在待掺入非天然氨基酸的位置处引入琥珀密码子5.分离表达高水平(大于0.5-20pg/细胞/天)的靶基因产物的稳定的克隆6.在存在允许在该琥珀密码子处掺入的非天然氨基酸的情况下,使该细胞系生长。根据此实施例,避免了高水平表达抑制型tRNA并且阻止了琥珀压制相关的细胞毒性。在此类方法中,表达tRNA适合地是在阻抑型启动子例如包含四环素应答元件(TetO或TtA)的H1以及U6启动子的控制下进行的(赫罗尔德(Herold(2008))。本发明的另一个方面是已根据前述方法中的一个制备的稳定化的细胞系。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系能够表达PylRS、tRNAPyl,并且该真核细胞系适于掺入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,该方法包括:(a)在一个或多个步骤中向真核细胞系中引入编码PylRS以及tRNAPyl的基因,其中tRNAPyl是在阻抑型启动子控制下,并且使得PylRS以及tRNAPyl稳定表达于所述细胞系中;(b)在存在阻抑物的情况下,培养所得的细胞系,使得tRNAPyl的表达受到阻抑,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系能够表达PylR、tRNAPyl以及包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质,该方法包括:(a)在一个或多个步骤中向真核细胞系中引入编码PylRS以及tRNAPyl的基因,其中tRNAPyl是在阻抑型启动子控制下,并且使得PylRS以及tRNAPyl稳定表达于所述细胞系中;(b)在存在阻抑物的情况下,培养所得的细胞系,使得tRNAPyl的表达受到阻抑,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响;(c)将编码包含一个或多个非天然氨基酸的靶蛋白质的基因引入所述真核细胞系,以使得所述靶蛋白质稳定表达于所述细胞系中;并且(d)在不存在所述阻抑物的情况下,表达靶蛋白质,以使得tRNAPyl被表达。“诱饵蛋白质”途径根据可替代的实施例,提供了如下方法,其中tRNAPyl对细胞生活力和/或细胞生长的不利影响被减小或被消除的条件包括如下条件,其中包含一个或多个由琥珀密码子编码的非天然氨基酸的诱饵蛋白质也在诱导型启动子控制下通过所述细胞系表达。例如,该诱饵蛋白质是选自:绿色荧光蛋白、红色荧光蛋白质、黄色荧光蛋白、青色荧光蛋白、蓝色荧光蛋白、白蛋白、SEAP、肌动蛋白、b-2微球蛋白、谷胱甘肽-s-转移酶、IgG、或包含聚琥珀的肽。因此,将非天然氨基酸掺入蛋白质的方法可包括以下步骤:1.将在诱导型启动子的控制下的针对诱饵蛋白质的包含琥珀密码子的基因引入真核细胞中2.分离细胞池或克隆池,该细胞或克隆包含诱饵构建体并且在诱发时能够以高水平(大于0.1或大于1pg/细胞/天)表达此蛋白质3.在一个或多个质粒上将该RS以及tRNA引入这些细胞,并且针对包含该RS以及tRNA的克隆进行选择4.在存在非天然氨基酸的情况下,使用整合的诱饵构建体分离能够在所希望的位点以大于30%,例如大于40%或50%或60%或70%或80%或优选地大于90%的有效率掺入这些非天然氨基酸的克隆5.将包含琥珀密码子的靶基因在非天然氨基酸被掺入的位置引入到真核细胞中6.分离能够有大于1pg/细胞/天的靶蛋白质的表达水平的克隆7.在存在允许在琥珀密码子处掺入的非天然氨基酸的情况下使该细胞系生长,并且分离显示非天然氨基酸在所希望的位点的掺入效率大于30%,例如大于40%或50%或60%或70%或80%或优选地大于90%的细胞本发明的另一个方面是稳定的真核细胞系,该真核细胞系在诱导型启动子控制下表达PylRS、tRNAPyl以及诱饵蛋白质。根据此实施例,当开始表达靶蛋白质时,诱饵蛋白质的表达可被中断(例如通过去除针对该启动子的诱导剂)。适合的诱导型启动子系统包括有条件地活化的启动子以及启动子系统,例如四环素调控型启动子(TetO或tTA;TetOn以及TetOFF)、多西环素-诱导型(TRE)启动子、cAMP诱导型启动子、糖皮质激素活化型启动子系统、IPTG诱导型启动子(lac)、Cd2+或Zn2+诱导型启动子(金属蛋白启动子)、干扰素依赖性启动子(例如鼠MX启动子)、HIVLTR启动子(Tat)、DMSO诱导型启动子(GLVP/TAXI,蜕皮激素)、以及雷帕霉素诱导型启动子(CID)。根据此实施例,当开始使用重组系统表达靶蛋白质时,诱饵蛋白质的表达可被中断(例如通过去除该基因)。适合的系统包括靶向重组系统,例如Cre/lox、基于phi31C的整合系统、以及Flp-FRT重组技术或通过插入盒的同源重组。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系能够表达PylRS、tRNAPyl以及包含一个或多个由无义密码子编码的非天然氨基酸的诱饵蛋白质,并且该真核细胞系适于掺入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,该方法包括:(a)在一个或多个步骤中将编码pylRS、tRNApyl以及所述诱饵蛋白质的基因引入真核细胞系,并且使得PylRS、tRNAPyl以及该诱饵蛋白质稳定表达于所述细胞系中;(b)培养所得的细胞系,该培养是在tRNAPyl仅由所述细胞系表达同时诱饵蛋白质也由所述细胞系表达的条件下,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响。本发明的另一个方面是用于产生稳定的真核细胞系的方法,该真核细胞系能够表达PylRS、tRNAPyl、诱饵蛋白质以及包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质,该方法包括:(a)在一个或多个步骤中将编码PylRS、tRNApyl以及诱饵蛋白质的基因引入真核细胞系,所述诱饵蛋白质在诱导型启动子控制下表达,并且使得PylRS、tRNAPyl以及诱饵蛋白质稳定表达于所述细胞系中;(b)在tRNAPyl仅由所述细胞系表达同时诱饵蛋白质也由所述细胞系表达的条件下,培养所得的细胞系,从而减小tRNAPyl对细胞生活力和/或细胞生长的不利影响;(c)向所述真核细胞系中引入编码包含一个或多个非天然氨基酸的靶蛋白质的基因,以使得所述靶蛋白质稳定表达于所述细胞系中;并且(d)表达靶蛋白质而不表达诱饵蛋白质。通用本发明的另一个方面是通过上述方法中的任何一个获得的或可通过其获得的稳定的真核细胞系。本发明的另一个方面是通过如下方法获得的或可通过其获得的稳定的真核细胞系,该方法是根据修饰该系统的上述方法(即,使用诱饵蛋白质、诱饵氨基酸、阻抑型启动子/诱导型启动子,在引入Pyl-tRNA等之前引入包含无义密码子的靶基因)的两个或更多个的组合。本发明的另一个方面是用于制备包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的方法,该方法包括在存在该一个或多个非天然氨基酸的来源的情况下,培养如上所述的稳定的真核细胞系。本发明的另一个方面是用于制备包含一个或多个由琥珀密码子编码的非天然氨基酸的靶蛋白质的方法,该方法包括向如上所述的稳定的真核细胞系中引入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,使得靶蛋白质稳定表达于所述细胞系中,并且在存在该一个或多个非天然氨基酸的来源的情况下并且在不存在诱饵蛋白质的表达的任何诱导剂的情况下表达所述靶蛋白质。本发明的另一个方面是用于制备包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的方法,该方法包括向如上所述的稳定的真核细胞系中引入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,使得靶蛋白质稳定表达于所述细胞系中,并且在存在该一个或多个非天然氨基酸的来源的情况下并且在不存在任何诱饵氨基酸的情况下表达所述靶蛋白质。本发明的另一个方面是用于制备包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的方法,该方法包括向如上所述的稳定的真核细胞系中引入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,使得靶蛋白质稳定表达于所述细胞系中,并且在存在该一个或多个非天然氨基酸的来源的情况下并且在不存在tRNAPyl的表达的任何阻抑物的情况下表达所述靶蛋白质。本发明的另一个方面是用于制备包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的方法,该方法包括向如上所述的稳定的真核细胞系中引入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,使得靶蛋白质稳定表达于所述细胞系中,并且在存在该一个或多个非天然氨基酸的来源的情况下表达所述靶蛋白质。本发明的另一个方面是用于制备经化学修饰的靶蛋白质的方法,该方法包括制备包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质,这包括向如上所述的稳定的真核细胞系中引入编码包含一个或多个由无义密码子编码的非天然氨基酸的靶蛋白质的基因,使得靶蛋白质稳定表达于所述细胞系中,在存在该一个或多个非天然氨基酸的来源的情况下表达所述靶蛋白质,并且化学修饰所得的靶蛋白质。用于根据本发明使用的细胞系本发明涉及稳定的真核细胞系。适合地这些细胞系是哺乳动物细胞系。更优选地,该细胞系是CHO细胞系,但也可以是HEK293、PERC6、COS-1、HeLa、VERO、或小鼠杂交瘤细胞系。另外的实例是COS-7以及小鼠骨髓瘤细胞系。CHO以及HEK293细胞系是特别适合的。本发明的某些元件可在无细胞表达系统的背景下使用,其中获得自宿主细胞的合成反应溶菌产物包括合成多肽所需的至少一种组分。该合成反应溶菌产物获得自细菌或真核细胞。优选地,该合成反应溶菌产物获得自真核细胞,更优选地,获得自兔网织红细胞或小麦胚芽。该无细胞表达系统能够表达本发明的WTPylRS和tRNApyl,其中tRNApyl被引入用于获得合成反应溶菌产物的具有本发明的DNA构建体的细胞中。适于用在本发明中的无细胞表达系统描述于,例如WO201008110、WO2010081111、WO2010083148,将其通过引用以其全文结合在此。根据本发明待表达于细胞系中的PylRS如此处使用的,pylRS涉及氨基酰基tRNA合成酶,该氨基酰基tRNA合成酶会氨酰基化适合的tRNA分子与吡咯赖氨酸或其衍生物。适合地,本发明的pylRS是正交于真核细胞中的吡咯赖氨酰-tRNA合成酶,这些真核细胞源自于产甲烷古菌物种-即,它是产甲烷古菌物种中的野生型或是其突变体。优选地,本发明的pylRS是源自于以下各项中的一个的吡咯赖氨酰-tRNA合成酶:马氏甲烷八叠球菌(SEQIDNO.1、SEQIDNO.2、SEQIDNO.14、SEQIDNO.15、SEQIDNO.16、)、巴氏甲烷八叠球菌(SEQIDNO.3、SEQIDNO.17)、哈夫尼脱亚硫酸菌(SEQIDNO.4、SEQIDNO.18)、埃斯蒂瓦兰斯甲烷八叠球菌(SEQIDNO.5、SEQIDNO.19)、布氏甲烷八叠球菌(Methanosarcinaburtonii)(SEQIDNO.6、SEQIDNO.20)、嗜热甲烷八叠球菌(SEQIDNO.7、SEQIDNO.21)、织里甲烷咸菌(SEQIDNO8、SEQIDNO.22)、伊夫氏甲烷盐菌(Methanohalobiumevastigatum)(SEQIDNO.9、SEQIDNO.23)、马希甲烷嗜盐菌(SEQIDNO.10、SEQIDNO.24)、吉布森脱硫肠状菌(SEQIDNO.11、SEQIDNO.25)、梅里迪维脱硫芽孢弯曲菌(Desulfosporosinusmeridei)(SEQIDNO.12、SEQIDNO.26)以及乙酸氧化脱硫肠状菌(SEQIDNO.13、SEQIDNO.27)。最优选地,本发明的pylRS是源自于马氏甲烷八叠球菌(SEQIDNO.1)的吡咯赖氨酰tRNA合成酶(pylRS)本发明的pylRS可以是野生型合成酶。可替代地,本发明的pylRS可在一个或多个位置突变,例如以增加其催化活性和/或修饰其针对底物氨基酸的选择性(柳泽2008)。优选地,本发明的pylRS可在相应于SEQIDNO.1的Tyr384位置突变或是其等价物。最优选地,Tyr384突变为苯基丙氨酸(SEQIDNO.2)。在一个实施例中,本发明的pylRS可在一个或多个位置突变以修饰其底物特异性并允许(或改善)吡咯赖氨酸类似物的掺入另外的突变体PylRS酶描述于WO09038195以及WO2010114615中,将每个文献以其全文通过引用掺入在此。根据本发明待表达于细胞系中的tRNApyl待与本发明的pylRS组合表达的tRNApyl具有反密码子以及互补于琥珀无义密码子UAG的三级结构,以起抑制型tRNA的作用。可构建互补于其他无义密码子例如UGA,蛋白石;UAA,赭石密码子的人工tRNA,以起抑制型tRNA的作用。因此,应当理解,尽管本发明通过提及使用用于编码nnAA的琥珀密码子并且讨论了琥珀压制的背景而充分描述并且示例,该琥珀密码子可被其他无义密码子例如蛋白石或赭石密码子代替,并且会被预期以相同方式起作用。然而优选使用琥珀密码子。工程化tRNApyl序列以最佳化在真核细胞系中的表达已在通过引用并入在此结合的WO2007099854中描述。WO2007099854尤其提供了包括以下各项的DNA构建体:源自于古细菌的tRNA基因(优选tRNApyl)、位于所述tRNA基因的3’的转录终止子序列、位于所述tRNApyl基因5’的通过RNA聚合酶II或III诱导转录的启动子序列,例如U1snRNA启动子或U6snRNA启动子。优选地,本发明的tRNApyl是源自于以下细菌菌株中的一个的tRNApyl:马氏甲烷八叠球菌(SEQIDNO28)、巴氏甲烷八叠球菌(SEQIDNO26)、哈夫尼脱亚硫酸菌(SEQIDNO30)、埃斯蒂瓦兰斯甲烷八叠球菌(SEQIDNO27)、布氏甲烷八叠球菌((SEQIDNO29)、或甲烷八叠球菌。更优选地,本发明的tRNApyl是源自于马氏甲烷八叠球菌(SEQIDNO28)的tRNApyl在本发明的一个实施例中,该tRNApyl表达于真核细胞,优选地动物细胞,更优选哺乳动物细胞中。在优选的实施例,天然缺少用于表达于真核细胞中的启动子元件的tRNApyl是在外部真核启动子下表达的。在一个特别优选的实施例中,该外部启动子是U6启动子。在一个特别优选的实施例中,该外部启动子是H1启动子。在另外的实施例中,该携带该tRNApyl基因的质粒包含该tRNApyl基因3’处的转录终止序列、该tRNApyl基因5’位置处的U6启动子序列以及该启动子区5’位置处的CMV增强子区。在特别优选的实施例中,携带该tRNApyl基因的质粒的插入片段具有SEQIDNO35。在本发明的一个实施例中,该外部启动子是阻抑型启动子在优选的实施例中,该阻抑型启动子选自:包含允许阻抑此启动子的元件的H1,例如TetO(H1/TetO;SEQIDNO31),或包含允许阻抑表达的元件的人类U6snRNA的启动子(例如U6/TetO)。应理解,如果指示靶基因的末端的终止密码子是琥珀密码子,并且意在使用琥珀密码子来编码该nnAA,那么该终止密码子会被改变为其他终止密码子(例如赭石或蛋白石)。如果意在使用其他无义密码子来编码该nnAA,则加以必要的变通同样适用。用于使用编码pylRS以及tRNAPyl的基因转化真核细胞系的载体本发明提供了用于tRNAPyl于真核细胞中有效表达的质粒。优选地,该tRNApyl表达质粒包括在U6启动子下SEQID28)的tRNA基因的多个重复。更优选地,该tRNApyl表达质粒包括在U6启动子下SEQID28)的tRNA基因的串联重复根据本发明,该PyltRNA基因以及该PylRScDNA是通过相同或不同的质粒携带的。在一个实施例中,该tRNApyl基因以及该PylRScDNA存在于相同的质粒上。在一个实施例中,该tRNApyl基因以及该PylRScDNA存在于不同的质粒上用于使用编码靶蛋白质的基因转化真核细胞系的载体本发明提供了包括本发明的核苷酸序列的载体,可任选地该核苷酸序列可操作地连接到启动子序列。在本发明中利用的载体包括:pJTI-Fast-DEST(生命技术公司(Lifetechnologies))、pSelect-Blasti以及pSelect-Zeo载体(英杰公司)、pENTRP5-P2载体(生命技术公司)、pOptivec(生命技术公司)、pFUSE-CHIg-hG1(英杰公司)以及pFUSE-CHLIg-hK(英杰公司)。适合的启动子的实例包括但不限于:CMV启动子,SV40大型T启动子(LargeTpromoter)、EF1α启动子、MCK启动子、以及LTR启动子。稳定细胞系的构建以及稳定克隆的选择诸位发明人已观察到,稳定表达针对位点特异性nnAA掺入所必需的元件的细胞系需要顺序引入用于表达该系统的不同元件(pylRS、tRNA、以及靶标)的质粒,每项跟随一个选择步骤和一个分选步骤(克隆步骤),以鉴别具有高活性的稳定的克隆。在本发明的一个实施例中,获得了稳定细胞,该细胞表达针对以下的所有元件:有效nnAA引入以充当引入靶基因的起点并且随后分离在所希望位置修饰的靶蛋白质。适合地,此途径包括反复选择方法,借此针对tRNA的表达盒首先被引入该宿主细胞中,并且凭借由该载体赋予的抗生素抗性选择包含这些构建体的细胞池。接着,通过引入编码来自维多利亚多管发光水母的绿色荧光蛋白质(GFP)的报告基因构建体选择存活的细胞,该构建体包含琥珀终止密码子,通过瞬时转染中断其开放阅读框。该选择方法在于:鉴别在琥珀压制时产生全长GFP的那些克隆,荧光是通过流式细胞术定量的。以此使用荧光激活细胞分选从此群体分离高功能细胞。将最好的克隆繁殖并且随后用该tRNA的另外拷贝转染,随后反复上述的选择方法。继续该方法,引入包含用于表达PylRS的盒以及多个拷贝的tRNA的整合表达构建体,直至获得每个组分表达以及测试琥珀压制的最佳水平。掺入通过琥珀密码子编码的非天然氨基酸可在使用本发明的细胞系制备的蛋白质中掺入一个或多个nnAA。适合地将一个nnAA掺入到蛋白质链中。在该蛋白质是抗体的情况下,可将一个nnAA掺入到该轻链或该重链或两者中。在其他实施例中,可将多于一个例如高达四个例如两个(或许三个)nnAA掺入到蛋白质链中。适合地,所有掺入的nnAA是相同的。可由琥珀密码子编码用于掺入到靶蛋白质中的非天然氨基酸通过引用结合在此的WO2007/130453中披露,使用非天然氨基酸以允许将多个部分共轭到肽上。如此处使用的,“非天然氨基酸”是指除硒代半胱氨酸以及二十种遗传编码的α-氨基酸之外的任何氨基酸、修饰的氨基酸、或氨基酸类似物,这二十种遗传编码的α-氨基酸是:丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酰胺、谷氨酸、甘氨酸、组氨酸、异亮氨酸、亮氨酸、赖氨酸、蛋氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸、缬氨酸。α-氨基酸的通用结构是通过式I说明的:典型地非天然氨基酸是具有式I的任何结构,其中该R基团是除了在这二十个天然氨基酸中使用的一种之外的任何取代基。参见,例如,任何针对这二十种天然氨基酸的结构的生物化学文本,例如L.史赛克(Stryer),第3版.1988,弗里曼和公司(FreemanandCompany),纽约的生物化学(Biochemistry)。注意,此处披露的非天然氨基酸可以是除了以上的二十种α-氨基酸之外的天然发生的化合物。因为此处披露的非天然氨基酸典型地仅在侧链上不同于这些天然氨基酸,这些非天然氨基酸与其他氨基酸(例如,天然的或非天然的)形成酰胺键,以与它们在天然发生的蛋白质中形成所相同的方式。然而,这些非天然氨基酸具有使它们区别于天然氨基酸的侧链基团。例如,式I中的R可任选地包括烷基-、芳基-、芳基卤化物、乙烯基卤化物、烷基卤化物、乙酰基、酮、氮杂环丙烷、腈、硝基、卤化物、酰基-、酮基-、叠氮基-、羟基-、肼、氰基-、卤基-、酰肼、烯基、炔基、醚、硫醚、环氧化物、砜、硼酸、硼酸酯、硼烷、苯基硼酸、硫醇、硒代-、磺酰基-、硼酸盐(borate)、硼酸(boronate)、磷、膦酰基、膦、杂环-、吡啶基、萘基、苯甲酮、环烷烃(例如约束环例如环辛炔、环烯烃例如降冰片烯、反式环烯烃、环丙烯)、四嗪、吡喃酮、硫酯、烯酮、亚胺、醛、酯、硫代酸、羟胺、氨基、羧酸、α-酮羧酸、α或β不饱和酸和酰胺、乙醛酰酰胺、或有机硅烷基团、或类似物,或其任意组合。除了包含新的侧链的非天然氨基酸之外,非天然氨基酸也可任选地包括修饰的骨架结构,例如,如通过式II以及III的结构说明的:其中Z典型地包括OH、NH2、SH、NH2O--、NH--R'、R'NH--、R'S--,或S--R'--;可以相同或不同的X以及Y典型地包括S、N、或O,并且可任选地相同或不同的R以及R'典型地选自上述针对具有式I的非天然氨基酸的R基团的组分的相同清单连同氢或(CH2)x或这些天然氨基酸侧链。例如,此处披露的非天然氨基酸可任选地包括如通过式II以及III说明的氨基或羧基基团上的取代。此类型的非天然氨基酸包括但不限于.α.-羟基酸、.α.-硫代酸.α.-氨基硫代羧酸盐、或.α.-.α.-双取代的氨基酸,侧链对应于例如这二十种天然氨基酸或对应于非天然侧链。它们也包括但不限于.β.-氨基酸或.γ.-氨基酸,例如取代的.β.-丙氨酸以及.γ.-氨基丁酸。此外,在.α.-碳上的取代或修饰可任选地包括L或D异构体,例如D-谷氨酸盐、D-丙氨酸、D-甲基-O-酪氨酸、氨基丁酸等等。其他结构性替代物包括环状氨基酸,例如脯氨酸类似物连同3-、4-、6-、7-、8-、以及9元环脯氨酸类似物。一些非天然氨基酸,例如芳基卤化物(p-溴-苯基丙氨酸、p-碘苯基丙氨酸),提供了通用的与乙炔(ethyne或acetylene)反应的钯催化的交联反应,这些反应允许在芳基卤化物和很多偶联伴侣之间形成碳-碳、碳-氮以及碳-氧键。例如,许多非天然氨基酸基于天然氨基酸,例如酪氨酸、谷氨酰胺、苯基丙氨酸等等。多种示例性非限制性非天然氨基酸的结构提供于US2003/0108885A1中,参见附图,例如,图29、30、以及31,将该文献的全部内容通过引用结合在此。氨基酸类似物的其他实例包括(但不限于)赖氨酸或吡咯赖氨酸氨基酸的非天然类似物,其包括以下官能团中的一个:烷基、芳基、酰基、叠氮基、腈基、卤基、肼、酰肼、羟基、烯基、环烯烃、炔基、环炔烃(例如约束环例如环辛炔、环链烯烃例如降冰片烯、反式环链烯烃、环丙烯)、芳基卤化物、乙烯基卤化物、烷基卤化物、氮杂环丙烷、硝基、羟基、醚、环氧化物、乙烯基醚、甲硅烷基烯醇醚、硫醇、硫醚、磺酰胺、磺酰基、砜、硒基、酯、硫代酸、硼酸、硼酸酯、硼烷、膦酰基、膦、杂环、吡啶基、萘基、苯甲酮、四嗪、吡喃酮、烯酮、亚胺、醛、羟胺、酮、硫酯、酯、硫代酸、有机硅烷基团、氨基、可光激活的交联剂;旋转标记的氨基酸;荧光氨基酸;与另一个分子发生共价或非共价相互作用的氨基酸;结合金属的氨基酸;包含金属的氨基酸;放射性氨基酸;光笼困的(photocaged)氨基酸;可光异构化的氨基酸;包含生物素或生物素类似物的氨基酸;糖基化的或碳水化合物修饰的氨基酸;包含酮的氨基酸;包括聚乙二醇的氨基酸;包括聚醚的氨基酸;重原子取代的氨基酸;可化学裂解的或可光裂解的氨基酸;具有伸长的侧链的氨基酸;包含有毒基团的氨基酸适合用于本发明的方法中的非天然氨基酸也包括具有荧光氨基酸的那些,例如包含萘基或丹酰基或7-氨基香豆素或7-羟基香豆素侧链的那些,可光裂解的或可光异构化的氨基酸,例如包含偶氮苯或硝基苄基Cys、Ser或Tyr侧链的那些,p-羧基-甲基-L-苯基丙氨酸、高谷氨酰胺、2-氨基辛酸、p-叠氮基苯基丙氨酸、p-苯甲酰苯基丙氨酸、p-乙酰基苯基丙氨酸、m-乙酰基苯基丙氨酸、2,4-二氨基丁酸(DAB)等等。本发明包括以上各项的未保护的和乙酰基化的形式。(还参见,例如,WO03/031464A2,标题为“肽的重构和糖共轭”;以及美国专利号6,331,418,标题为“糖组合物,用于合成它们的方法和装置”;唐(Tang)以及蒂雷尔(Tirrell),美国化学会志(J.Am.Chem.Soc.)(2001)123:11089-11090)以及唐(Tang)等人,德国应用化学国际版(Angew.Chem.Int.Ed.),(2001)40:8,通过引用将所有这些以其全文结合在此)。在本发明中,可利用以上具有式IV的非天然氨基酸(nnAA)以产生蛋白质。在一个实施例中,附接至酰胺基部分的X基团可以是烷基叠氮化物、烷氧基叠氮化物、烷氧基环氧化物、烷基-炔烃、烷氧基炔烃、烷氧基链烯烃、烷基-链烯烃、烷基链、烷基环己烯、烷基环炔烃、烷氧基环链烯烃、烷氧基环炔烃、酰胺基环炔烃、酰胺基环链烯烃、反环式环链烯烃、环丙烯、四嗪、吡喃酮、降冰片烯、芳基叠氮化物、叠氮基、羟基胺、肼、乙烯基卤化物、芳基卤化物、四嗪、吡喃酮、亚胺、硼酸酯或酸、氰基基团、羰基基团,例如醛或酮。在优选的实施例中,以上通用结构的非天然氨基酸(nnAA)可具有从氨基酸末端至相对末端处的酰胺基基团的1-12亚甲基基团的烷基链。优选地,以上通用结构的非天然氨基酸(nnAA)可包含作为该连体结构的部分的环烷烃以及芳族环。在一个实施例中,以上通用结构的非天然氨基酸(nnAA)与吡咯赖氨酰tRNA合成酶(PyRS)以及tRNApyl相互作用。所述氨基酸包括:(S)-2-氨基-6-((2-叠氮基乙氧基)羰基氨基)己酸(Lys-叠氮化物)、(S)-2-氨基-6-((丙-2-炔基氧基)羰基氨基)己酸(Lys-炔烃)、S)-2-氨基-6((2-氧代-2-苯基乙酰胺)己酸、S)-2-氨基-6((2-氧代-2-丙酰胺)己酸、(2S)-2-氨基-6-({[(2-叠氮基环戊基)氧基]羰基}氨基)己酸、(2S)-2-氨基-6-({[(2-乙炔基环戊基)氧基]羰基}氨基)己酸、(2S)-2-氨基-6-{[(环辛-2-炔-1-基氧基)羰基]氨基}己酸、(2S)-2-氨基-6-({[2-(环辛-2-炔-1-基氧基)乙氧基]羰基}氨基)己酸、(2S)-2-氨基-6-[({双环[2.2.1]庚-5-烯-2-基氧基}羰基)氨基]己酸、(2S)-2-氨基-6-[({双环[2.2.1]庚-5-烯-2-基甲氧基}羰基)氨基]己酸、(2S)-2-氨基-6-{[({4-[(6-甲基-1,2,4,5-四嗪-3-基)氨基]苯基}甲氧基)羰基]氨基}己酸、(2S)-2-氨基-6-({[(4E)-环辛-4-烯-1-基氧基]羰基}氨基)己酸、(2S)-2-氨基-6-{[(环丙-2-烯-1-基氧基)羰基]氨基}己酸、(2S)-2-氨基-6-{[(环丙-2-烯-1-基甲氧基)羰基]氨基}己酸、(2S)-2-氨基-6-{[(3-叠氮基丙基)氨基甲酰基]氧基}己酸、(2S)-2-氨基-6-{[(丁-3-炔-1-基氧基)羰基]氨基}己酸、(2S)-2-氨基-6-(2-叠氮基乙酰胺基)己酸、(2S)-2-氨基-6-(3-叠氮基丙酰胺基)己酸、(2S)-2-氨基-6-(5-叠氮基戊酰胺基)己酸。该nnAA将是pylRS的底物。适合地,本发明的nnAA源自于赖氨酸。通过引用结合在此的WO2010139948描述了用于本发明的感兴趣的若干nnAA,具体是以下的赖氨酸衍生物:(S)-2-氨基-6((丙-2-炔基氧基)羰基氨基)己酸(Lys炔烃):(S)-2-氨基-6((2叠氮基乙氧基)羰基氨基)己酸(Lys叠氮化物):其他适合的nnAA是:(S)-2-氨基-6((2-氧代-2-苯基乙酰胺)己酸:(S)-2-氨基-6((2-氧代-2-丙酰胺)己酸:(S)-2-氨基-6-(乙酰胺)己酸:(S)-2-氨基-6-(烯丙氧基羰基氨基)己酸:(2S)-2-氨基-6-({[(2-叠氮基环戊基)氧基]羰基}氨基)己酸:(2S)-2-氨基-6-({[(2-乙炔基环戊基)氧基]羰基}氨基)己酸:(2S)-2-氨基-6-{[(环辛-2-炔-1-基氧基)羰基]氨基}己酸:(2S)-2-氨基-6-({[2-(环辛-2-炔-1-基氧基)乙氧基]羰基}氨基)己酸:(2S)-2-氨基-6-[({双环[2.2.1]庚-5-烯-2-基氧基}羰基)氨基]己酸:(2S)-2-氨基-6-[({双环[2.2.1]庚-5-烯-2-基甲氧基}羰基)氨基]己酸:(2S)-2-氨基-6-{[({4-[(6-甲基-1,2,4,5-四嗪-3-基)氨基]苯基}甲氧基)羰基]氨基}己酸:(2S)-2-氨基-6-({[(4E)-环辛-4-烯-1-基氧基]羰基}氨基)己酸:(2S)-2-氨基-6-{[(环丙-2-烯-1-基氧基)羰基]氨基}己酸:(2S)-2-氨基-6-{[(环丙-2-烯-1-基甲氧基)羰基]氨基}己酸:(2S)-2-氨基-6-{[(3-叠氮基丙基)氨基甲酰基]氧基}己酸:(2S)-2-氨基-6-{[(丁-3-炔-1-基氧基)羰基]氨基}己酸:(2S)-2-氨基-6-(3-叠氮基丙酰胺基)己酸:(2S)-2-氨基-6-(5-叠氮基戊酰胺基)己酸:(2S)-2-氨基-6-(戊-4-烯酰胺基)己酸:另外的nnAA包括:(2S)-2-氨基-6-{[(3-叠氮基丙基)氨基甲酰基]氧基}己酸、(2S)-2-氨基-6-{[(3-叠氮基丙基)氨基甲酰基]氧基}己酸、(2S)-2-氨基-6-{[(丙-2-炔-1-基)氨基甲酰基]氧基}己酸、(2S)-2-氨基-6-{[(丁-3-炔-1-基)氨基甲酰基]氧基}己酸、(2S)-2-氨基-6-{[(丙-2-烯-1-基)氨基甲酰基]氧基}己酸、(2S)-2-氨基-6-{[(丁-3-烯-1-基)氨基甲酰基]氧基}己酸。适合地,本发明的nnAA源自于(2S)-2-氨基-6-羟基己酸。例如:(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸:(2S)-2-氨基-6-{[(3-叠氮基丙基)氨基甲酰基]氧基}己酸:(2S)-2-氨基-6-{[(丙-2-炔-1-基)氨基甲酰基]氧基}己酸:(2S)-2-氨基-6-{[(丁-3-炔-1-基)氨基甲酰基]氧基}己酸:(2S)-2-氨基-6-{[(丙-2-烯-1-基)氨基甲酰基]氧基}己酸:(2S)-2-氨基-6-{[(丁-3-烯-1-基)氨基甲酰基]氧基}己酸:另外的适于用在本发明中的非天然氨基酸类似物是具有式V的结构的吡咯赖氨酸类似物其中Z=化学键、CH2、CH-NH2、CH-OH、NH、O、S或CH-NH2;b是0或整数1-7;并且FG=叠氮化物、链烯烃、炔烃、酮、酯、芳基或环炔烃。在式V中,当FG表示芳基时,实例是芳族卤化物,例如4-卤代苯基,例如4-碘苯基。部分Z(CH2)bFG可以,例如,表示CO-芳基,例如CO-苯基或–CO烷基,例如-COMe。具有式V的示例性化合物如下:(2S)-2-氨基-6-{[(2-叠氮基乙氧基)羰基]氨基}己酸(2S)-2-氨基-6-{[(丙-2-炔-1-基氧基)羰基]氨基}己酸(2S)-2-氨基-6-{[(丙-2-烯-1-基氧基)羰基]氨基}己酸(2S)-2-氨基-6-(3-叠氮基丙酰胺基)己酸(2S)-2-氨基-6-(戊-4-烯酰胺基)己酸(S)-2-氨基-6((2-氧代-2-苯基乙酰胺)己酸(S)-2-氨基-6((2-氧代-2-丙酰胺)己酸(2S)-2-氨基-6-(2-叠氮基乙酰胺基)己酸适合用作本发明中的非天然氨基酸的可替代的吡咯赖氨酸类似物具有式VI的结构:其中Z=CH2、CH-NH2、CH-OH、NH、O或S;FG=叠氮化物、链烯烃、炔烃、酮、酯、芳基或环炔烃;并且b=1-4的整数。在式VI中,当FG表示芳基时,实例是芳族卤化物,例如4-卤代苯基,例如4-碘苯基。具有式VI的示例性化合物是:(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸(2S)-2-氨基-6-{[(丙-2-炔-1-基)氨基甲酰基]氧基}己酸(2S)-2-氨基-6-{[(丙-2-烯-1-基)氨基甲酰基]氧基}己酸在式V和VI的结构中,当FG表示链烯烃时,它适合地表示–CH=CH2或–CH=CH-CH3,优选地–CH=CH2。在式V和VI的结构中,当FG表示炔烃时,它适合地表示–C≡CH或–C≡C-CH3,优选地–C≡CH。在式V和VI的结构中,当FG表示酮时,它适合地表示–C(=O)-CH3或–C(=O)-CH2-CH3,优选地–C(=O)-CH3。在式V和VI的结构中,当FG表示酯时,它适合地表示–C(=O)-O烷基,例如–C(=O)-O甲基。在式V和VI的结构中,当FG表示芳族卤化物时,它适合地表示被卤素尤其碘取代的苯基(例如4-碘-苯基)。在式V和VI的结构中,当FG表示环炔烃时,它适合地表示环辛炔,例如环辛-4,5-炔。有利地,本发明的具有式V和VI的nnAA已显示具有如由GFP测定证明的良好的掺入。在作为掺入测定的GFP测定中,式VI.1与式V.1具有相似水平的翻译能力。式V和VI两者易被修饰以掺入各种有用的官能团,这些官能团可被用于位点选择性翻译后修饰。炔烃和链烯烃易被掺入。可使用各种方法制造此处披露的吡咯赖氨酸类似物。一般这些反应条件可由本领域的普通技术人员确定。式V类似物可容易地通过将活化的羰基基团(例如氯甲酸酯)、活化羧酸酯、异氰酸酯、活化碳酸酯或磺酰卤化物添加到类型1的单保护的二氨基底物来制备,其中ɑ-氨基基团受保护基团(“PG”)例如Boc、Cbz、TFA、乙酰基或Fmoc基团(参见方案1)的保护。偶联的产物3可经受另外的修饰,例如用叠氮基亲核试剂置换卤化物以安置所希望的官能度。否则,将中间体3去保护以去除ɑ-氨基酸掩蔽基团,以给予所希望的式V类似物。方案1:通过将羟基氨基酸9共轭至具有活化羰基例如羧酸酯、异氰酸酯、酰基氯、活化碳酸酯或磺酰卤化物的底物来制备式VI类似物。偶联的产物11可经受另外的修饰,例如通过置换离去基团例如卤化物或活化醇来设置叠氮化物官能团。通过最终去保护以去除ɑ-氨基酸掩蔽基团来获得所希望的氨基酸类似物12。可按照方案1使用保护基团。参见方案2:以上提供的非天然氨基酸中的许多是可商购的,例如,来自西格马奥德里奇(SigmaAldrich)(USA)。可任选地如在US2004/138106A1(通过引用结合在此)的实例中提供的或使用本领域的普通技术人员已知的标准方法来合成不可商购的那些。关于有机合成技术,参见,例如,有机化学(OrganicChemistry),费斯山登(Fessendon)以及费斯山登(Fessendon),(1982,第二版,威拉德·格兰特出版社(WillardGrantPress),波士顿,马萨诸塞州(BostonMass.));高等有机化学(AdvancedOrganicChemistry),玛奇(March)(第三版,1985,约翰威立父子出版公司(WileyandSons),纽约);以及高等有机化学,凯里(Carey)以及松德贝里(Sundberg)(第三版,部分A以及B,1990,普莱南出版公司(PlenumPress),纽约),以及WO02/085923,将所有这些通过引用结合在此。本发明的其他nnAA可通过公开的方法合成。例如,(S)-2-氨基-6((丙-2-炔基氧基)羰基氨基)己酸以及S)-2-氨基-6((2叠氮基乙氧基)羰基氨基)己酸的合成公开于WO2010139948以及阮等人2009中。S)-2-氨基-6((2-氧代-2-苯基乙酰胺)己酸、S)-2-氨基-6((2-氧代-2-丙酰胺)己酸、(2S)-2-氨基-6-({[(2-叠氮基环戊基)氧基]羰基}氨基)己酸、(2S)-2-氨基-6-({[(2-乙炔基环戊基)氧基]羰基}氨基)己酸、(2S)-2-氨基-6-{[(环辛-2-炔-1-基氧基)羰基]氨基}己酸、(2S)-2-氨基-6-({[2-(环辛-2-炔-1-基氧基)乙氧基]羰基}氨基)己酸、(2S)-2-氨基-6-[({双环[2.2.1]庚-5-烯-2-基氧基}羰基)氨基]己酸、(2S)-2-氨基-6-[({双环[2.2.1]庚-5-烯-2-基甲氧基}羰基)氨基]己酸、(2S)-2-氨基-6-{[({4-[(6-甲基-1,2,4,5-四嗪-3-基)氨基]苯基}甲氧基)羰基]氨基}己酸、(2S)-2-氨基-6-({[(4E)-环辛-4-烯-1-基氧基]羰基}氨基)己酸、(2S)-2-氨基-6-{[(环丙-2-烯-1-基氧基)羰基]氨基}己酸、(2S)-2-氨基-6-{[(环丙-2-烯-1-基甲氧基)羰基]氨基}己酸披露于郝(Hao),Z.,化学通讯(Chem.Comm.),47,4502,2011,舒尔茨(Schultz)PG,等人,自然方法(Nat.Methods),4,239-244,2007,舒尔茨PG,等人,生物有机化学与医药化学快报(Bioorg.Med.Chem.Lett.),15,1521-1524,2005,迪特斯(Dieters)A.,等人,美国化学会志(J.Am.Chem.Soc.),125,11782-11783,2005,王(Wang),YS,等人,美国化学会志,134,2950-2953,2012,费克内尔(Fekner),T.,等人,德国应用化学英文国际版(AngewChemIntEdEngl)45,1633-1635,2009.,普拉斯(Plass),T.,等人德国应用化学英文国际版,51,4166-4170,2012,朗(Lang),K.美国化学会志,134,10317,2012以及戴瓦拉吉(Devaraj)NK,德国应用化学英文国际版,48,7013-7016,2009。具有掺入的非天然氨基酸的蛋白质的用途使用根据本发明的方法,具有掺入的非天然氨基酸的蛋白质可被用于制备官能化蛋白质共轭物。可被共轭至具有掺入的非天然氨基酸的蛋白质的分子包括(i)其他蛋白质,例如抗体,尤其单克隆抗体;以及(ii)聚合物,尤其PEG基团或其他可引起系统中半衰期延长的基团。此外,这些修饰的蛋白质可被共轭至药物或核苷酸,用于这些高效化合物的靶向递送。因此可被共轭至具有掺入的非天然氨基酸的蛋白质的其他分子包括(iii)细胞毒性剂以及(iv)药物部分。某些实施例的更多细节在以下抗体药物共轭的讨论中给出。常规地,非天然的氨基酸可包含独特的化学基团,该化学基团允许以靶向方式共轭而没有与其他氨基酸发生副反应的风险。例如非天然氨基酸方便地包含叠氮化物或炔烃基团,该叠氮化物或炔烃基团允许使用胡伊斯根(Huisgen)1,3-偶极环加成反应,与待被共轭的包含相应的炔烃或叠氮化物基团的分子发生反应。位点特异性共轭本发明的另一个方面是用于制备经化学修饰的靶蛋白质的方法,该方法包括根据本发明的方面所述的方法制备靶蛋白质并且化学修饰所得的靶蛋白质。本发明的优选的共轭化学包括正交于天然的二十个氨基酸的反应。此类反应不与天然的20个氨基酸相互作用或引起副反应,这20个氨基酸对于与该反应相关的官能团是特异性的。适合地,通过该nnAA,这些必需官能团被掺入到靶蛋白质中。此外,所述反应在对于该蛋白质是非破坏性的条件下进行,例如水性溶剂,pH值范围是该蛋白质可以接受的并且维持其溶解性,温度对该蛋白质不会导致有害影响。增加该蛋白质和该连接体之间的附接部分的稳定性可以是有利的。常规方法通过与马来酰亚胺反应形成硫醇醚,共轭至半胱氨酸的硫醇基团。该硫醇醚可经受逆反应,从该抗体释放连接体药物衍生物。在本发明的实施例中,叠氮化物和炔烃之间采用的共轭化学导致芳族三唑,该芳族三唑显著地更稳定,并且不容易发生可逆性。此外,该反应的产物、蛋白质和有效载荷之间的键应该是稳定的,等于或大于与常规键(酰胺、硫醇醚)相关的稳定性。虽然不是对共轭的妨碍,如果共轭反应可以在天然条件下完成的话通常是有利的,因为这将消除额外的重折叠处理步骤。用于产生本发明的共轭物的优选化学共轭包括:3+2炔烃-叠氮化物环加成;3+2双极环加成;基于钯的偶联,包括赫克反应;薗头反应;铃木反应;施蒂勒(Stille)偶联;桧山(Hiyama)/丹麦反应;烯烃复分解;狄尔斯-阿尔德(Diels-alder)反应;与肼、酰肼、烷氧基胺或羟基胺的羰基缩合;张力促进的环加成,包括张力促进的叠氮化物炔烃环加成;金属促进的叠氮化物炔烃环加成;电子促进的环加成;片段挤出环加成;链烯烃环加成,随后b-消除反应。根据一个优选的实施例,该掺入的氨基酸包含叠氮化物或炔烃基团,并且化学修饰的方法包括使所述叠氮化物或炔烃基团与包括炔烃或叠氮化物基团的试剂反应。设想的反应是胡伊斯根1,3-偶极环加成反应,其导致三唑键的产生。包括炔烃或叠氮化物基团的试剂可以是携带炔烃或叠氮化物基团(可任选地通过连接体)的蛋白质(例如抗体)或毒素或细胞毒性药物或适于半衰期延长的物质(例如PEG基团)。用在所述反应中的炔烃基团是例如,环辛炔,例如双环[6.1.0]壬-4-炔部分(BCN)。在变体反应中,该掺入的氨基酸包含叠氮化物或链烯烃基团,并且化学修饰的方法包括使所述叠氮化物或链烯烃基团与包括链烯烃或叠氮化物基团的试剂反应。包括链烯烃或叠氮化物基团的试剂可以是携带炔烃或链烯烃基团(可任选地通过连接体)的蛋白质(例如抗体)或毒素或适于半衰期延长的物质(例如PEG基团)。在实施例中,本发明的共轭化学被用于制备抗体药物共轭物。产物的化学修饰如在本文别处指示的,根据本发明的细胞系有用于产生包含掺入的非天然氨基酸的蛋白质。有用地,可在另外的化学反应中采用所述非天然氨基酸。本发明的另一个方面是用于制备经化学修饰的靶蛋白质的方法,该方法包括根据本发明的方面所述的方法制备靶蛋白质并且化学修饰所得的靶蛋白质。本发明的优选的共轭化学包括正交于天然的二十个氨基酸的反应。此类反应不与天然的20个氨基酸相互作用或引起副反应,这20个氨基酸对于与该反应相关的官能团是特异性的。此外,所述反应在对于该蛋白质是非破坏性的条件下进行,例如水性溶剂,pH值范围是该蛋白质可以接受的并且维持其溶解性,温度对该蛋白质不会导致有害影响。根据一个实施例,该掺入的氨基酸包含叠氮化物或炔烃基团,并且化学修饰的方法包括使所述叠氮化物或炔烃基团与包括炔烃或叠氮化物基团的试剂反应。设想的反应是胡伊斯根1,3-偶极环加成反应,其导致三唑键的产生。包括炔烃或叠氮化物基团的试剂可以是携带炔烃或叠氮化物基团(可任选地通过连接体)的蛋白质(例如抗体)或药物部分(例如毒素或细胞毒性药物)或适于半衰期延长的物质(例如PEG基团)。可任选地,该胡伊斯根1,3-偶极环加成反应可在Cu(I)催化作用的存在下进行。优选地,铜催化的环加成反应是在室温下,在水性溶液中,在半胱氨酸和三[(1-苄基-1H-1,2,3-三唑-4-基)甲基]胺(TBTA)的存在下进行。可替代地,该铜催化的环加成反应是在从4℃至50℃下,在水性溶液中,在抗坏血酸钠和三(3-羟丙基三唑基甲基)胺(THPTA)的存在下进行的。该反应还可在混合的水性/有机溶液中进行,其中有机组分由以下各项组成:DMSO、DMF、甲醇、乙醇、t-丁醇、三氟乙醇、丙二醇、乙二醇和己二醇。在变体反应中,该掺入的氨基酸包含叠氮化物或链烯烃基团,并且化学修饰的方法包括使所述叠氮化物或链烯烃基团与包括链烯烃或叠氮化物基团的试剂反应。包括链烯烃或叠氮化物基团的试剂可以是携带炔烃或链烯烃基团(可任选地通过连接体)的蛋白质(例如抗体)或毒素或适于半衰期延长的物质(例如PEG基团)。当多于一个nnAA被掺入到靶蛋白质(例如抗体)中时,该化学修饰可以相同或不同。例如如果两个nnAAs被掺入,一个可被修饰为共轭至药物部分,并且一个可被修饰为共轭至PEG部分。靶蛋白质靶蛋白质包括抗体,特别是单克隆抗体。本发明的抗体包括全长抗体和抗体片段,包括Fab、Fab2,以及单链抗体片段(scFv),针对TROP-2、SSTR3、B7S1/B7x、PSMA、STEAP2、PSCA、PDGF、RaSL、C35D3、EpCam、TMCC1、VEGF/R、连接蛋白-30、CA125(Muc16)、脑信号蛋白-5B、ENPP3、EPHB2、SLC45A3(PCANAP)、ABCC4(MOAT-1)、TSPAN1、PSGRD-GPCR、GD2、EGFR(Her1)、TMEFF2、CD74、CD174(leY)、Muc-1、CD340(Her2)、Muc16、GPNMB、Cripto、EphA2、5T4、间皮素、TAG-72、CA9(IX)、a-v-整合素、FAP、Tim-1、NCAM/CD56、α叶酸受体、CD44v6、硫酸软骨素蛋白聚糖、CD20、CA55.1、SLC44A4、RON、CD40、HM1.24、CS-1、β2微球蛋白、CD56、CD105、CD138、路易斯Y、GRNMP、Tomoregulin、CD33、FAP、CAIX、FasL受体、MMP底物金属蛋白酶。在本发明的优选实施例中,本发明的针对肿瘤靶的抗体共轭至选自以下项的蛋白质部分:免疫刺激的以及促凋亡的蛋白质,特别是免疫刺激物例如IL-1α、IL-1β、其他IL-1家族成员;白介素的任一种,包括但不限于IL-2、IL-4、IL-5、IL-6、IL-7、IL-12、IL-13、IL-15、IL-17家族、IL-18、IL-21、IL-22、IL-23、IL-28;或共刺激配体,例如B7.1以及B7.2、TACI。干扰素,例如类型IIFN家族(IFNα以及β以及λ)或类型IIIFNγ。造血生长因子,例如GM-CSF。趋化因子,包括CXCL-1、CXCL-2、CXCL-5、CXCL-6、CXCL-8、CXCL-9、CXCL-10、以及CXCL-11、CXCL-13、CCL-2、CCL-3、CCL-4、CCL-5、CCL-21、IP-10、嗜酸性粒细胞趋化因子(Eotaxin)、RANTES、PF4、GRO相关的肽、IL-8。促凋亡配体,例如TNF超家族的那些,包括FasL、TNF、PD-L1。抗微生物肽,例如α以及β防卫素以及组织蛋白酶抑制素(cathelicidin)LL37/hCAP18、富组蛋白、组织蛋白酶G、天青杀素、糜酶、来源于嗜酸性粒细胞的神经毒素、高泳动族1核内蛋白、HMGB1、乳铁蛋白。产生ROS以及RNS的酶,例如NADPH氧化酶(NOX)的成员、氧化氮合酶NOS,INOS)、中性粒细胞颗粒蛋白(包括蛋白酶,例如弹性蛋白酶以及组织蛋白酶)、天青杀素(也称作CAP37或HBP)、髓过氧化物酶、穿孔素、颗粒酶。在一个实施例中靶蛋白质是抗-Her-2抗体。在一个实施例中,靶蛋白质是抗-IL-6抗体。在一个实施例中,靶蛋白质是抗-PSMA抗体。在优选的实施例中,该抗-PSMA抗体是单链抗体(scfv)。在一个实施例中,靶蛋白质是FGF21,例如具有SEQIDNo62的序列或与其具有95%一致性(例如与其具有96%、97%、98%或99%一致性)的序列。序列鉴别是将全蛋白质作为比较窗口计算的。可使用常规的序列比较程序例如BLAST。在优选的实施例中,FGF21被修饰以在位置R131包含非天然氨基酸lys-叠氮化物或炔丙基赖氨酸(参见SEQIDNo.64),并通过三唑连接体共轭至PEG部分。诱饵氨基酸用在根据本发明的方法中的诱饵氨基酸是未掺入到延伸的蛋白质中的氨基酸衍生物。可替代地,诱饵氨基酸是掺入到延伸的蛋白质中但抑制蛋白质伸长的氨基酸衍生物。本发明的诱饵氨基酸具有通式VII:其中G=H、OH、-OCH3、OCH2CH3、O-C(=O)-CH3或NH-K-Q;X=化学键、CH2、S、O、NH、N-(C=O)-或CH-J;J=烷基、芳基、杂芳基或20个天然氨基酸中一个的侧链;Y=化学键、NH、O、S、CH2;Z=O、NH、CH2、S、CH-NH2;K=CO或SO2;a=0、1、2或3;b=0、1、2或3;Q=-H、C1-6烷基、芳基、杂芳基-OC1-6烷基、-OCH2芳基、-OCH2杂芳基、-C2-6烯基或–OC2-6烯基;并且R=C1-6烷基、C2-6烯基、-CH2芳基、C2-6炔基、C1-6卤代烷基或C1-6叠氮基烷基。在Q的定义内的示例芳基基团是苯基。示例R基团包括-CH2CH=CH2、-CH2CH2Cl、-CH2CH2N3、-CH2Ph、-C(CH3)3、-CH2CH2CH3、-CH2CH3、-CH3、-CH(CH3)2,-以及-CH2-C≡C-H。在Q的定义内的示例芳基基团是苯基。针对Q的示例基团包括H、-CH3、-Et、Ph、-OtBu、-OFmoc、-OBn、-OMe、-OEt以及-OCH2CH=CH2。在一个实施例中,K是CO。在另一个实施例中,K是SO2。适合地,Y表示NH、O或S,并且Z表示O、NH、CH2、S或CH-NH2,或者Y表示键、NH、O、S或CH2,并且Z表示O、NH或S。适合地,Y表示NH、O或S。适合地,Z表示NH、O或S。适合地,Y表示NH、O或S,并且Z表示NH、O或S。在一个实施例中,Y是NH,并且Z是O。在另一个实施例中,Y是O,并且Z是NH。当J表示20个天然氨基酸中一个的侧链时,实例包括半胱氨酸、丝氨酸、苏氨酸、天冬氨酸、谷氨酸、丙氨酸、苯丙氨酸、异亮氨酸、缬氨酸、酪氨酸和色氨酸的侧链。在实施例中,本发明的诱饵氨基酸是如下的氨基酸,该氨基酸是pylRS的底物,具有化学修饰的胺基团,例如具有式VIIA的N-酰化氨基酸:其中K是CO或SO2;Q=H、C1-6烷基、芳基、杂芳基–OC1-6烷基、-OCH2芳基、-OCH2杂芳基、-C2-6烯基或–OC2-6烯基。有利地,本发明的诱饵nnAA能够阻止由表达该PyltRNA引起的琥珀压制的毒性作用。通过使能够在存在琥珀抑制型tRNA的情况下终止该琥珀密码子处的蛋白质翻译,该诱饵阻止琥珀压制。适合地,具有式VII的诱饵氨基酸,其缺少传播多肽合成所必需的氨基末端基团,如在式VIIB中:其中G=H;a=4或5;并且R=C1-6烷基、C2-6烯基,-CH2芳基、C2-6炔基、C1-6卤代烷基或C1-6叠氮基烷基。在Q的定义内的示例芳基基团是苯基。当R表示C1-6叠氮基烷基时,适合地它表示C2-6叠氮基烷基,例如C2-4叠氮基烷基。示例R基团包括-CH2CH=CH2、-CH2CH2Cl、-CH2CH2N3、-CH2Ph、-C(CH3)3、-CH2CH2CH3、-CH2CH3、-CH3、-CH(CH3)2、以及-CH2-C≡C-H。在一个实施例中,a是4。在另一个实施例中,a是5。示例性的具有式VIIB的诱饵氨基酸是以下:6-{[(丙-2-烯-1-基氧基)羰基]氨基}己酸;5-{[(丙-2-烯-1-基氧基)羰基]氨基}戊酸;6-{[(2-氯乙氧基)羰基]氨基}己酸;6-{[(叔丁氧基)羰基]氨基}己酸;6-{[(丙-2-炔-1-基氧基)羰基]氨基}己酸;以及6-{[(2-叠氮基乙氧基)羰基]氨基}己酸基于实例12中的数据,该诱饵nnAA能够阻止由表达该PyltRNA引起的琥珀压制的毒性作用。通过使能够在存在琥珀抑制型tRNA的情况下终止该琥珀密码子处的蛋白质翻译,该诱饵阻止琥珀压制。诱饵蛋白质用在根据本发明的方法中的诱饵蛋白质是非靶的包含一个或多个由琥珀密码子编码的非天然氨基酸的良性蛋白质。本发明的诱饵蛋白质选自:绿色荧光蛋白、红色荧光蛋白、白蛋白、SEAP、肌动蛋白、b-2微球蛋白、谷胱甘肽-s-转移酶以及包含聚琥珀的肽。另外的实例是IgG。本发明的诱饵蛋白质适合地在诱导型启动子控制下,这些启动子选自有条件地活化的启动子以及启动子系统,例如四环素调控型启动子(TetO或tTA;TetOn以及TetOFF)、多西环素-诱导型(TRE)启动子、cAMP诱导型启动子、糖皮质激素活化型启动子系统、IPTG诱导型启动子(lac)、Cd2+或Zn2+诱导型启动子(金属蛋白质启动子)、干扰素依赖性启动子(例如鼠MX启动子)、HIVLTR启动子(Tat)、DMSO诱导型启动子(球蛋白质启动子球蛋白质LCR)、激素调节型启动子(GLVP/TAXI,蜕皮激素)、以及雷帕霉素诱导型启动子(CID)。PEG部分靶蛋白质可被共轭至PEG部分。PEG部分可被掺入到抗体药物共轭物中。该PEG部分可典型地具有在0.5kDa和40kDa例如5kDa和40kDa之间变化的分子量。更优选地,该PEG部分可具有20kDa左右的分子量。。此外,该PEG部分可具有从100-2000Da变化的分子量。PEG部分可以是直链或支链的或多臂的。该PEG部分可以用末端炔烃、叠氮化物、氰化物、环炔烃、链烯烃、芳基卤化物官能化。该PEG可以如下方式官能化,例如单官能化、同双官能化、异双官能化和多-同官能化。抗体药物共轭物(ADC)根据本发明的细胞系特别用于产生具有均质性质的抗体药物共轭物(由合成连接体共价结合到给定药物(典型地细胞毒性药物)或者蛋白质或PEG基团的重组的抗体),其中每抗体的药物(或其他共轭分子)的数目和那些药物在该抗体上的位置被明确地控制,从而获得包含掺入的非天然氨基酸的单克隆抗体并且通过正交化学位点特异性地共轭至携带药物部分(或其他共轭分子)的连接体。适合地,本发明提供了用以获得ADC的方法,包括以下步骤:1.将一个或多个携带编码全长抗体的DNA序列的质粒引入本发明的稳定细胞系中,借此将终止密码子引入到该序列内的特异位置处2.对修饰的在所希望的一个或多个位置处具有装入的非天然的氨基酸(nnAA)的抗体进行纯化。3.通过正交化学使被修饰以包括如下官能团的细胞毒素-连接体衍生物与包含互补的反应性基团的修饰的抗体发生反应,该官能团互补于装入该抗体中的nnAA4.纯化所得到的ADC因此,本发明还提供了ADC,借此该抗体组分已被修饰以在所希望的位置掺入具有独特反应性官能团的非天然氨基酸,借此此类官能团允许共轭至药物部分(或蛋白质或PEG基团)。在实施例中,本发明提供了包括抗-Her-2抗体的抗体共轭物,该抗-Her-2抗体通过包括三唑部分的连接体而共轭至一个或多个(例如一个、两个、三个或四个,优选地一个或两个,尤其一个)选自蛋白质、药物和PEG部分的部分。具体言之,该三唑部分可以通过非天然氨基酸的侧链中的叠氮化物或炔烃部分和附接至蛋白质、药物或PEG部分的炔烃或叠氮化物部分的反应形成,该非天然氨基酸被掺入到抗-Her-2抗体的序列中。在一个实施例中,该三唑部分是通过在Cu(I)催化作用的条件下,非天然氨基酸的侧链中的叠氮化物或炔烃部分和附接至蛋白质、药物或PEG部分的炔烃或叠氮化物部分的反应形成,该非天然氨基酸被掺入到抗-Her-2抗体的序列中。Cu(I)催化作用是通过使用天然Cu(I)来源例如碘化铜、溴化铜、氯化铜、硫醇铜、氰化铜来完成。Cu(I)种类还可以通过使用铜(II)来源和还原剂原位产生。铜(II)来源可以是硫酸铜、氯化铜(II)、或乙酸铜。该还原剂可以是抗坏血酸钠、二硫苏糖醇、TCEP、b-巯基乙醇、肼、羟基胺、亚硫酸氢钠、胱胺、半胱氨酸适合地,Cu(I)催化的环加成是在配体的存在下进行的,以稳定存在于反应起始时或通过用抗坏血酸钠还原Cu(II)来源例如硫酸钠原位产生的Cu(I)种类,包括TBTA、THPTA、邻二氮杂菲衍生物、吡啶基甲亚胺衍生物、二亚乙基三胺、联吡啶衍生物、TMEDA、N,N-联(2-吡啶基甲基)胺(BPMA)衍生物、N,N,N’,N’-四(2-吡啶基甲基)乙二胺(TPEN)衍生物、三烷基胺(例如三乙胺、二异丙基乙胺)、HEPES和MES。在另一个实施例中,抗体共轭物包括抗体,该抗体通过包括三唑部分的连接体而共轭至选自药物以及PEG部分的一个或多个部分,其中该三唑部分是通过被掺入该抗体的序列中的非天然氨基酸的侧链中的叠氮化物部分和附接至药物或PEG部分的炔烃部分的反应形成,并且其中该炔烃部分是环辛炔部分。在另一个实施例中,抗体共轭物包括抗体,该抗体通过包括三唑部分的连接体而共轭至选自药物以及PEG部分的一个或多个部分,其中该三唑部分是通过被掺入该抗体的序列中的非天然氨基酸的侧链中的炔烃部分和附接至药物或PEG部分的叠氮化物部分的反应形成,并且其中该炔烃部分是环辛炔部分。该环辛炔部分可以是,例如,双环[6.1.0]壬-4-炔部分。适合地,掺入到该抗体的序列中的非天然氨基酸是pylRS的非天然氨基酸底物,特别是非天然赖氨酸类似物,例如(S)-2-氨基-6((2-叠氮基乙氧基)羰基氨基)己酸。抗体在本发明中,ADC包括使用全长抗体连同抗体片段,例如但不限于Fab、Fab2、以及单链抗体片段。适于共轭至细胞毒素的抗体包括靶向以下各项的那些:抗-Her2、抗-IL-6、TROP-2、SSTR3、B7S1/B7x、PSMA、STEAP2、PSCA、PDGF、RaSL、C35D3、EpCam、TMCC1、VEGF/R、连接蛋白-30、CA125(Muc16)、脑信号蛋白-5B、ENPP3、EPHB2、SLC45A3(PCANAP)、ABCC4(MOAT-1)、TSPAN1、PSGRD-GPCR、GD2、EGFR(Her1)、TMEFF2、CD74、CD174(leY)、Muc-1、CD340(Her2)、Muc16、GPNMB、Cripto、EphA2、5T4、间皮素、TAG-72、CA9(IX)、a-v-整合素、FAP、Tim-1、NCAM/CD56、α叶酸受体、CD44v6、硫酸软骨素蛋白聚糖、CD20、CA55.1、SLC44A4、RON、CD40、HM1.24、CS-1、β2微球蛋白、CD56、CD105、CD138、路易斯Y、GRNMP、Tomoregulin、CD33、FAP、CAIX、FasL受体、MMP底物金属蛋白酶。在优选实施例中,本发明的抗体是IgG类型的。在本发明的特别优选实施例中,该抗体被修饰以包括一个或多个非天然氨基酸,其中此类非天然氨基酸在IgG免疫球蛋白之中的位置是保守的并且选自SEQIDNo82的位置K157(表示针对IgG的重链的保守恒定区,对应于SEQIDNo46和75的抗-Her2抗体的位置274)以及SEQID82的位置T242(对应于SEQIDNo46和75的抗-Her2抗体的位置359)以及IgG的轻链的框架区中的位置D70和L81(遵循卡巴特编号并对应于SEQIDNo52和79的D70和L81)。为清楚起见,在以下氨基酸背景下发现D70:sgsrsgtdftltisslq,并且在以下氨基酸背景下发现E81:sslqpedfatyycqq。感兴趣的一种具体的抗体是抗-Her-2抗体。该抗-Her2-抗体可以,例如具有SEQIDNo52的轻链序列或与其具有95%(例如96%、97%、98%或99%)或更多序列一致性并且具有相同CDR的衍生物,以及SEQIDNo46的重链序列或与其具有95%(例如96%、97%、98%或99%)或更多序列一致性并且具有相同CDR的衍生物。序列鉴别是将全抗体(但排除CDR)作为比较窗口计算的。可使用常规的序列比较程序例如BLAST。此文献中所述的mAb序列与赫塞汀的序列具有高度相似性。此处利用的mAb序列是通过将赫塞汀中发现的抗原结合位点序列置入种系IgG1中产生的。针对Her2的细胞外结构域的小鼠抗体4D5的可变区是使用重叠寡聚物通过基因合成产生的并被克隆到穿梭载体中。然后这些可变区被接枝到由pFUSE-CHIg-hG1以及pFUSE-CHLIg-hK(英杰公司)编码的人框架以产生小鼠-人类杂交体。序列比较显示,构建的抗体相对于赫塞汀具有六个氨基酸取代。这些对应于5个重链位置以及一个轻链位点。这些位点中没有一个对应于CDR区域或邻近这些CDR的位点。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的重链序列的位置274处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的轻链序列的位置70处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的重链序列的位置274处,并且也在所述抗-Her2-抗体的每条轻链的轻链序列的位置70处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的重链序列的位置359处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条轻链的轻链序列的位置81处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的重链序列的位置274处,并且也在所述抗-Her2-抗体的每条轻链的轻链序列的位置81处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的重链序列的位置359处,并且也在所述抗-Her2-抗体的每条轻链的轻链序列的位置70处。根据实施例,用于共轭的非天然氨基酸在所述抗-Her2-抗体的每条重链的重链序列的位置359处,并且也在所述抗-Her2-抗体的每条轻链的轻链序列的位置81处。感兴趣的另一种具体的抗体是抗-PSMA抗体,尤其scfv。该抗-PSMA抗体可以,例如,具有SEQIDNo58的scfv序列或与其具有95%(例如96%、97%、98%或99%)或更多序列一致性并且具有相同CDR的衍生物。序列鉴别是将全抗体(但排除CDR)作为比较窗口计算的。可使用常规的序列比较程序例如BLAST。在具体实施例中,抗-PSMAscfv被修饰以在位置117(SEQID60)处包含非天然氨基酸lys-叠氮化物。所述scfv可以,例如,被共轭至MMAF-缬氨酸-胍氨酸-p-氨基-苯甲酰基-碳酸酯-环炔烃衍生物位点特异性修饰抗体用于产生ADC在本发明中,选择用于将nnAA掺入该抗体的共轭位点包括以下步骤:位点的最初选择是使用计算机模拟预测方法进行,将该抗体的三维结构、其功能性域以及在该抗体的结构或功能上起作用的关键氨基酸残基考虑在内。然后将选择的位点针对其物理-化学特性以及稳定性进行筛选。适合地,选择最佳共轭位点的标准包括以下:优选的位点是:该抗体的结合位点的远端的残基;表面/溶剂暴露的残基(以增强接近于共轭物形成并使能够有效地形成共轭物);根据经验发现允许有效的琥珀压制的位点;根据经验发现保持所表达的蛋白质以及共轭物的稳定性的位点。避免位点是:对于功能(例如FcRN结合、Fcγ相互作用)重要的残基、已知对于折叠或结构(例如Cys,脯氨酸)重要的氨基酸残基遵循以上标准概要已鉴别了人类IgG1中的六个位点。这些包括四个重链位置(T114、K274、K288以及T359)以及两个轻链位点(D70以及E81)。HCK274、K288以及T359;并且LCD70、L81显示有效地将nnAA掺入并且使能够形成共轭物。连接体根据本发明,该靶蛋白质或抗体可被直接连接到该蛋白质或药物部分或PEG部分或通过连接体或间隔子另外连接。本发明的连接体可以是可裂解的或非可裂解的。因此,本发明提供了抗体共轭物,其中该包括三唑部分的连接体凭借在该连接体中存在包含裂解位点的间隔子而是可裂解连接体。该裂解位点可以是酶促不稳定裂解位点。酶促不稳定裂解位点的实例是由酶组织蛋白酶B识别的缬氨酸-瓜氨酸肽的掺入并且在该瓜氨酸C-末端裂解该肽。在一个实施例中,该抗体共轭物的该连接体包括三唑部分,不是可裂解连接体。可裂解连接体的使用是通过对在其靶内待释放的处于不变状态的细胞毒素的需要来驱动的。这是通过细胞毒素示例的,例如单甲基auristateE<(佩蒂特(Pettit)1997,森特2003)>。以可裂解连接体释放的机制可以是化学例如酸不稳定性,或通过在该连接体中包含可裂解肽的酶促。该机制也可以是通过光或其他辐射来源或化学引发剂例如氟外部引发的。非可裂解连接体不须从该细胞毒素去除以在治疗过程中实现所希望的效价或细胞杀伤作用。因此,抗体被内在化并在溶酶体内被还原为其氨基酸组分,释放该药物-连接体。它是不需要另外的释放以成为高效的这种化合物。非可裂解连接体不具有用于释放完整的细胞毒素的内在机制,替代地,它们依赖于其内含物在细胞毒素框架上的良性状态。非可裂解连接体可具有多个变化的结构,从相对简单到更复杂的实体。另外,以此方式定义连接体,其中它们附接该细胞毒素以及抗体两者。对于常规的途径,此包括针对附接至半胱氨酸硫醇(马来酰亚胺)或赖氨酸胺(活化酸)的化学。本发明的连接体掺入炔烃或叠氮化物基团。适合地,本发明的非可裂解连接体包括在一末端附接至抗体的功能性把手(Y),桥接ADC的两个组分并且提供了附接至抗体以及至药物所需的官能团的间隔子以及用于偶联至药物的互补官能团(X)。适合地,优选的功能性把手是那些化学部分,它们是装入靶蛋白质的非天然氨基酸的官能团的互补的反应性配偶。该分子的间隔子部分是非官能性化学桥,其包含附接至抗体以及至药物所需的两个互补性官能团。在该连接体的这个实施例中,此间隔子不具有裂解位点。在本发明的优选实施例中,该功能性把手(Y)包括炔烃基团。优选地,该炔烃可以是末端炔烃、内部炔烃、环状炔烃以及甲硅烷基-保护的炔烃。优选地,该内部炔烃将包含邻近该炔烃的吸电子基团。优选地,该环状炔烃将是包含在7、8或9元环内的炔烃。这些吸电子基团包括卤素,例如氟、溴氯以及碘。另外的吸电子基团包括羟基、醚、缩醛、缩酮、酮、醛、羧酸、酯、腈、硝基、酰胺。更优选地,该环将被包括在双环的环系统中,其中该8元环被稠合到具有3、4、5、或6个原子的其他环,如在以下中描述的,例如范代尔夫特(vanDelft),F.,应用化学(Angew.Chem.)(49/1/-5/2012)M.D.贝斯特(Best),生物化学(Biochemistry)2009,48,6571-6584;E.M。塞拉顿(Sletten),C.R.贝尔托西(Bertozzi),应用化学2009,121,7108-7133;应用化学国际版(Angew.Chem.Int.Ed.)2009,48,6974-6998.;J.A.普雷舍尔(Prescher),C.R.贝尔托西,自然化学生物学(Nat.Chem.Biol.)2005,1,13-21,J.A.科德丽(Codelli),J.M.巴斯金(Baskin),N.J.阿加德(Agard),C.R.贝尔托西,美国化学会志(J.Am.Chem.Soc.)2008,130,11486-11493。将这些文献通过引用结合在此。特别优选的环状炔烃是在US7,807,619以及USSN12/049,034(通过引用结合在此)中所述的。特别优选的双环炔烃是在WO2011/136645(通过引用结合在此)中所述的双环壬炔:在一个实施例中,本发明的非可裂解连接体可包含作为在该抗体附接位点的功能性把手(Y)的链烯烃。适合地,该链烯烃可以是单、双、三或四取代的。适合地,该链烯烃可作为环的部分掺入。在优选的实施例中,该链烯烃可以是3-12元环的部分。在另外的优选的实施例中,该链烯烃可以是双环的环系统的部分,例如降冰片烯、双环呋喃或双环吡咯系统。优选地,在该抗体附接位点处的功能性把手(Y)包括乙烯基卤化物。优选地,该乙烯基卤化物包括在Z或Y位置或两个位置处的卤化物,例如氟、氯、溴、或碘。此外,该乙烯基卤化物可以是末端的,其中该R-基团是氢。该乙烯基卤化物基团也可包含在该R位置处的另外的取代,包括烷基以及芳基基团、羰基基团。优选地,该乙烯基卤化物是环状化合物的部分。更优选地,该乙烯基卤化物是具有3、4以及5个原子的环的部分。在一个实施例中,在该抗体附接位点处的功能性把手(Y)包括在LG位置被甲硅烷基基团以及卤化物或三氟甲磺酸盐、甲苯磺酸盐或甲磺酸盐取代的反应性芳族环。在另外的实施例中,在该抗体附接位点处的功能性把手(Y)包括在该末端的反应性叠氮化物基团。适合地,本发明的可裂解连接体包括用以在一个末端附接至抗体的功能性把手(Y),间隔子以及用于偶联至该药物的互补官能团(X)。适合地,本发明的ADC的可裂解连接体包括裂解位点。适合地,该裂解位点可以是酶促地、化学地或外部地引发的。在一个实施例中,该裂解位点位于该药物附接位点。在可替代实施例中,该裂解位点在该药物附接位点。在实施例中,该可裂解连接体包括在该抗体附接位点处的具有炔烃的功能性把手(Y)。优选地,该炔烃可以是末端炔烃、内部炔烃、环状炔烃以及甲硅烷基-保护的炔烃。优选地,该内部炔烃将包含邻近该炔烃的吸电子基团。这些吸电子基团包括卤素,例如、氟、溴氯以及碘。另外的吸电子基团包括羟基、醚、缩醛、缩酮、酮、醛、羧酸、酯、腈、硝基、酰胺。优选地,该环状炔烃将是包含在7、8或9元环内的炔烃。更优选地,该环将被包括在双环的环系统中,其中该8元环被稠合到具有3、4、5、或6个原子的其他环。在可替代实施例中,该裂解位点以及间隔子在顺序上是颠倒的,其中该裂解位点在该药物附接位点。在实施例中,本发明的可裂解连接体可包含作为在该抗体附接位点的功能性把手(Y)的链烯烃。适合地,该链烯烃可以是单、双、三或四取代的。适合地,该链烯烃可作为环的部分掺入。在优选的实施例中,该链烯烃可以是3-12元环的部分。在另外的优选的实施例中,该链烯烃可以是双环的环系统的部分,例如降冰片烯、双环呋喃或双环吡咯系统。优选地,在该抗体附接位点处的功能性把手(Y)包括乙烯基卤化物。优选地,该乙烯基卤化物包括在Z或Y位置或两个位置处的卤化物,例如氟、氯、溴、或碘。此外,该乙烯基卤化物可以是末端的,其中该R-基团是氢。该乙烯基卤化物基团也可包含在该R位置处的另外的取代,包括烷基以及芳基基团、羰基基团。优选地,该乙烯基卤化物是环状化合物的部分。更优选地,该乙烯基卤化物是具有3、4以及5个原子的环的部分。在可替代实施例中,该裂解位点以及间隔子在顺序上是颠倒的,其中该裂解位点在该药物附接位点。在一个实施例中,在该抗体附接位点处的功能性把手(Y)包括在LG位置被甲硅烷基基团以及卤化物或三氟甲磺酸盐、甲苯磺酸盐或甲磺酸盐取代的反应性芳族环。在可替代实施例中,该裂解位点以及间隔子在顺序上是颠倒的,其中该裂解位点在该药物附接位点。在另外的实施例中,在该抗体附接位点处的功能性把手(Y)包括在该末端的反应性叠氮化物基团。在可替代实施例中,该裂解位点以及间隔子在顺序上是颠倒的,其中该裂解位点在该药物附接位点。在本发明的实施例中,可裂解的以及非可裂解连接体两者的间隔子部分可以是结构多样的,并且包括烷基链、烷基环、芳族环、苯胺衍生物(包括p-氨基-苄基碳酸酯)、链烯烃、聚合物例如聚乙二醇。在本发明的优选实施例中,连接体是由在一个末端的环炔烃构成,用于通过叠氮化物-炔烃环加成附接至抗体。附接至环炔烃是碳链,然后其附接至缬氨酸-瓜氨酸肽。瓜氨酸的C-末端偶联至p-氨基-苯甲酰基氨基甲酸酯(PABC)。进而其连接到MMAF的N-末端。此缬氨酸-瓜氨酸肽是通过酶组织蛋白酶B识别的,该组织蛋白质酶B在瓜氨酸c-末端裂解该肽。在裂解之后,该p-氨基苯甲酰基经受消除反应以挤出CO2以及MMAF基团。因此,整个环-炔烃-val-cit-PABC组合是可裂解连接体。-在本发明的实施例中,在引发剂引发后,连接体从ADC释放药物适合地,可在靶细胞附近或内部发现引发剂。优选地,引发剂是在细胞内发现。适合地,细胞内的引发剂包括酶促引发剂。适合地,酶促裂解位点包括通过胞内酶特异性识别的氨基酸序列。本发明的优选酶促裂解位点是组织蛋白酶(缬氨酸-瓜氨酸)以及弗林蛋白酶(Furin)(Arg-N-Arg-Arg),可替代地,化学引发剂是在靶细胞内发现。适合地,化学引发剂包括以下化学部分的酸水解,包括酯、酰胺、缩醛、缩酮、腈、醚裂解、氨基甲酸酯、脲、磺酰胺、磺酰基、氧硫基、磷烷酰胺、氨基磷酸酯、烯胺、亚胺、甲硅烷基醚、原酸酯、硼酸酯。可替代地,化学引发剂也可包括以下化学部分的还原,包括二硫化物、氟加成至甲硅烷基基团、反向环加成和反向迈克尔加成。可替代地,从连接体释放药物可通过细胞外刺激(例如暴露于特定波长的辐射)实现。药物部分本发明的药物部分,例如细胞毒素药物部分,包括小分子、天然产物、合成上衍生的药物、蛋白质例如免疫毒素和放射性核素。在实施例中,该药物部分是阿里他汀部分,例如阿里他汀或其衍生物,例如单甲基阿里他汀E(MMAE)(威多廷(Vedotin))或单甲基阿里他汀F(MMAF)、阿里他汀F(AF)、鹅膏蕈碱、紫杉醇和阿霉素。其他药物部分包括美登素、紫杉醇、阿霉素和免疫毒素,例如外毒素或bouganin,连同放射性核素,例如碘-131、钇-90、钐-135和锶-89,还可以将其掺入到有机分子中(参见例如:MMAE:森特(Senter),PE等人,血液(BLOOD),102,1458-1465.MMAF:森特,PE等人,生物共轭化学(Bioconj.Chem.)2006,17,114-124.美登素:路易斯-飞利浦(Lewis-Phillips)GD,癌症研究(CancerRes.),63,9280-9290,2008.Bouganin:MacDonaldGC等人,免疫疗法杂志(J.Immunotherapy),32574-84,2009)。最适合地,该药物部分是选自阿霉素、紫杉醇和阿里他汀部分的部分。盐类此处所述的氨基酸、氨基酸衍生物、诱饵氨基酸以及吡咯赖氨酸类似物可任选地以盐的形式采用。任何此类盐形成本发明的部分。羧酸的盐可包括与基团1和基团2金属,尤其可溶盐例如钠和钾盐形成的盐。胺的盐可包括与弱酸和强酸,例如HCl、HBr或乙酸形成的盐。实例实例1:稳定细胞系的产生产生能够位点特异性地将nnAA整合到靶蛋白质中的平台细胞系需要分步骤地构建稳定表达pylRS以及该pyltRNA的细胞系。这通过顺序引入pylRS/tRNA表达元件以及反复的选择步骤以鉴别高功能细胞而完成(图1)。将包含以下各项的质粒转染到DG44-CHO细胞中:九个拷贝U6-pyltRNA表达盒,连同编码人INF基质连接区的序列(pSB-9xtRNA-MARS,借此U6以SEQID32定义,tRNA序列以SeqID28定义),介导核中染色质的组织并在基因表达的调控中发挥作用并且通过复制增强这些元件的稳定性的序列元件(克拉尔(Klar)2005;衡(Heng)2004;皮夏切克(Piechaczek)1999),并且在补充以HT补充物的ProCHO4培养基中进行选择,该HT补充物包含0.1mM次黄嘌呤以及0.016mM胸苷,8mM谷氨酰胺,5ug/mL杀稻瘟菌素(ProCHO4-C)。然后使用GFP报告基因测定与细胞分选针对tRNA功能对细胞进行细胞选择。简而言之,将细胞用编码FLAG标记的pylRS(SEQID2)的pJTI-R4PylRSeGFPY40以及包含琥珀密码子替代编码位置40处的酪氨酸的密码子的报告子eGFP(eGFPY40,SEQID38)进行转染,并且将细胞暴露于2mMnnAAALOC(N-烯丙基氧基羰基-L-赖氨酸)持续14h。使用BDFACSAriaII细胞分选仪将显示最高水平的荧光的30,000个细胞收集到新鲜的ProCHO4-C培养基中并扩展。此群体的细胞表示包含pyltRNA活性的分选池。为了测试该分选池是否显示比亲本(预分选池)改善的功能,将两个群体都用编码野生型eGFP(SEQID37)的pJTI-R4PylRSeGFPY40GFP对照(pTracerEF/HisA;生命技术公司,被修饰以包含F64L以及S65T突变)瞬时转染。使经转染的细胞在包含2mMALOC的ProCHO4-C培养基中生长24h并且使用Accuri流式细胞仪分析,并且在这些以及对照细胞(图2A)中对荧光水平进行定量。这些数据显示,与亲本菌株或未经转染的对照比较,经分选的细胞群体在存在nnAA的情况下具有更高的琥珀压制效率。这个中间体细胞群体称作DG44-CHO-191。虽然该DG44-CHO-191细胞能够琥珀压制,其效率受到限制,具有少于43%琥珀压制(基于GFPY40)。显示tRNA的水平是琥珀压制的效率的限制因子。因此,将经分选的DG44-CHO-191细胞用pSZ-9xtRNA转染并且在DMEM-BZ(DMEM(生命技术公司),2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%胎牛血清,HT补充物,5ug/ml杀稻瘟素,0.5mg/mL博莱霉素)中选择细胞。将存活的细胞池(称作该DG44-CHO-200-12)以及DG44-CHO-191细胞用pJTI-R4PylRSeGFPY40或pTracer转染。包含tRNA表达盒的另外拷贝的细胞证明了增加的eGFPY40依赖性荧光并且从而证明了琥珀压制效率(图2B)。这些数据显示,该分步的反复选择和细胞分选方法得到对具有改善的功能的细胞的鉴别。理解到tRNA是该系统的限制组分,并且另外增加了pyltRNA的表达水平并且从而这一细胞群体的琥珀压制效率,使DG44-CHO-200-12细胞经受细胞分选以分离具有高琥珀压制能力的细胞s。此处,将DG44-CHO-200-12细胞(包含pSB-9x-MARS以及pSZ-9x)用pJTI-R4-pylRS-eGFPY40瞬时转染并且使细胞在包含2mMALOC的培养基中生长。将显示最高的1%荧光水平的7,000个细胞使用BDFACSAriaII细胞分选仪分离并在DMEM-BZ中繁殖。这得到称作DG44-CHO-208-2的细胞池。该平台细胞系的完成需要该稳定地引入用于pyrlRS的表达的盒。因此,将208-2细胞用携带编码SEQIDNo2(Y384F突变体)或SEQID1(WT)的pylRS的cDNA序列的pMOAV2或pMOAV2-puro转染,并在包含0.5mg/mL潮霉素(DMEM-HBZ)的DMEM-BSD-Zeo或包含7.5ug/ml嘌呤霉素(DMEM-PBZ)的DMEM-BZ中选择转化体,以产生经选择的细胞池,称作DG44-CHO-211-1(hygro)或DG44-CHO-211-2(puro)。培养抗生素抗性细胞并用编码eGFP报告子构建体的pENTR-P5-P2eGFPY40转染,并将细胞在存在2mMALOC的情况下培养1小时20min,并且随后将显示高荧光水平的细胞使用细胞分选进行分离。此处,分离211-1的1331个细胞(来自1,712,332个事件)以及211-2的1169个细胞。将称作DG44-CHO-223-1或DG44-CHO-223-2的经分选的群体培养于DMEM-HBZ或DMEM-PBZ中。为了确定对包含pylRS的群体的分选是否改善了琥珀压制效率,用编码GFP的包含中断其开放阅读框的琥珀密码子(P2-P5eGFPY40)、eTracer的报告子质粒对DG44-CHO-223-1和其亲本细胞系DG44-CHO-211-1进行瞬时转染,或不经转染。将经转染的细胞用2mMALOC孵育28h并且通过流式细胞术利用Accuri流式细胞仪进行荧光定量(图2C)。虽然DG44-CHO-211-1以及DG44-CHO-223-1显示等效转染能力(eTracer对照),但DG44-CHO-223-1比亲本细胞系显示超过5-倍多的eGFPY40依赖性荧光。此结果指示,分选细胞使得能够分离高活性琥珀压制细胞并且分离有效平台细胞系。接着,将该平台细胞系用于开发包含编码待用nnAA修饰的蛋白质的稳定整合基因靶的表达细胞系。将该DG44-CHO-223-2细胞系用包含用于表达针对人类IL-6、具有在重链cDNA的位置K274处的琥珀密码子的IgG的基因的pOtivec-28D2amb274质粒转染。为了这样做,通过枝接产生针对人细胞因子IL-6的抗体,这是将针对人细胞因子IL-6的兔抗体的可变区通过PCR扩增并且克隆到载体pFUSE-CHIg-hG1(重链,SEQID40)以及pFUSE-CHLIg-hK(轻链,SEQID44)(英杰公司)中枝接到人框架,以产生兔-人杂交体,如在通过引用以其全文结合在此的WO2012032181中所述。在该重链恒定区的位置K274处通过定点诱变(SEQID42)引入琥珀密码子。通过DNA测序鉴别包含该琥珀密码子的克隆。为了产生此IgG的整合构建体,将针对该重链的启动子和ORF通过PCR扩增,并通过限制酶消化并连接到pOptivec(生命技术公司(LifeTechnologies))中而克隆。将该轻链和tRNA的单拷贝通过两步骤PCR方法使用重叠寡聚物进行连接,并克隆到可用位点到包含该重链的pOptivec质粒中。将所得到的载体通过转染引入到平台细胞系DG44-CHO-223-2中,并通过在缺少次黄嘌呤以及胸苷的生长培养基,即DMEM-HT(DMEM,2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%透析的胎牛血清,5ug/ml杀稻瘟素,0.5mg/mL博莱霉素,0.75ug/mL嘌呤霉素)中培养生长选择细胞。该Optivec载体也包含针对二氢叶酸还原酶(DHFR)的基因,这使得DG44CHO细胞能够在缺少HT补充物的培养基中并且在存在甲氨蝶呤的情况下生长。将细胞进一步在缺少DMEM-HT并且包含10nM、50nM、以及100nM甲氨蝶呤(MTX)的培养基中进行选择。收获活的细胞,并在具有之前概述的所使用的抗生素浓度的一半的相同培养基中将这些细胞以50细胞/孔分布于96孔盘中。在生长培养基中不存在nnAA的情况下,pylRS/tRNA对是无活性的并且琥珀压制不发生。以此,表达截短的IgG重链并且分泌到生长培养基中。之后10-12天,将各孔针对生长进行监测并且将ELISA测定用于鉴别包含表达高水平的截短的IgG的菌落的孔。为了这样做,将ELISA板用磷酸盐缓冲盐水(PBS)中的1ug/mL3xFLAG-IL-6-Avi在4℃涂覆1h或过夜。之后在水中洗涤并在包含1%BSA的PBS中阻滞,将15ul的表达培养基用35ul的包含0.1%脱脂乳的PBS稀释,并且添加到每个孔中在室温持续1h。将孔在水中洗涤若干次,并且将50ul的1:10,000稀释的该第二抗体共轭至辣根过氧化物酶(抗-人类H+L-HRP;杰克逊实验室(JacksonLaboratories))在室温持续1h。然后洗涤各孔并将50ul的SureblueReserveTMB(KPL)添加到每个孔。之后5-10分钟,添加0.1NH2SO4以停止该反应,并使用读板仪在450nM波长定量显色。繁殖包含表达高水平的截短的IgG的细胞的孔并扩展。这个测定导致了对显示截短的IgG高表达的七个克隆的鉴别。为了确定所分离的克隆是否显示有效的琥珀压制,将这些克隆暴露于2mMALOC中,并通过ELISA测量全长IgG的表达水平。简而言之,将山羊人抗-FC抗体(杰克森实验室)用于特异性地捕获全长IgG,并且不是截短的IgG。这七个测试的克隆之一显示高水平全长表达(3F2,SEQID42)。为了证明琥珀压制的效率,利用3F2克隆用于表达和纯化IgG。在组织培养烧瓶中在包含50nMMTX的培养基中将该3F2克隆培养到90%汇合,并且将细胞在表达培养基(DMEM,2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%低IgG胎牛血清,5ug/ml杀稻瘟素,0.5mg/mL博莱霉素,0.75ug/mL嘌呤霉素)中与2mMlys-叠氮化物(nnAA)孵育。允许细胞表达抗体持续7天并收获培养基。在蛋白质A柱上捕获来自表达上清液的抗体并用PBS洗涤。将结合的蛋白在50mM甘氨酸pH3.0中洗脱并且将包含IgG的峰级分用PBS透析。将包含lys-叠氮化物nnAA的经纯化的抗体称作AzAb。将代表样本重悬浮于SDS-PAGE装载缓冲液中,并且在还原和非还原条件下分别通过SDS-PAGE解析0.5ug和1ug,并且用考马斯亮蓝染色(图3A)。为了证明所表达的产物包含nnAA(lys-叠氮化物),将所表达的蛋白质用100倍过量的包含环状炔烃官能团的20KDa-PEG在室温孵育4h。将等量的起始材料以及PEG-IgG共轭物通过SDS-PAGE解析并通过考马斯染色可视化(图3B)。该PEG改变了该共轭物的分子量,导致凝胶迁移率的阻滞。当在变性以及还原条件下解析该反应混合物时,观察到仅被设计为包含该nnAA整合位点(在位置274)的IgG的重链显示胶迁移性位移。相比之下,该轻链似乎未被该共轭反应改变。这些数据证明,所表达的蛋白质包含特异性地修饰的部分,并且这些共轭条件对该重链是特异性的。为了进一步证明共轭物观察到的迁移性位移表示IgG抗-IL-6AzAb(对照IgG)的PEG化,将用于该共轭反应的起始材料以及该共轭IgG结合到蛋白质A。将该结合材料用PBS洗涤以去除未共轭的PEG,并且将蛋白质用2%SDS洗脱。然后将此材料通过SDS-PAGE在还原条件下进行解析,并且将蛋白质通过考马斯亮蓝染色可视化,碘染色以可视化PEG,并且使用抗-人类FC特异性抗体(杰克森实验室)进行蛋白质印迹以检测该重链(图3C)。这些数据显示,在该重链上特异性地形成共轭物,并且分子量增加是归因于形成了与PEG的共轭物。将表达针对IL-6的抗体的第二个克隆进行鉴别并且平行(7B1,SEQID42)表征为如以上指示的产生的关于针对her2/neu的抗体的克隆(3E9,SEQID48),包含在上述的相同位置编码到该重链中的琥珀密码子(K274,SEQID48)。对这些细胞系表达水平的进行定量,并且在表达培养基中在存在lys-叠氮化物的情况下确定每个细胞产率(图3D)。这些数据证明本方法的可应用性在于表达包含nnAA的不同抗体。-实例2:琥珀压制相关的毒性在该平台细胞系分离的过程中,诸位发明人观察到,随着增加量的pylRS/tRNApyl被引入以改善该系统的效率,这些细胞的生活力恶化。具体言之,发现增加的tRNApyl水平对琥珀压制效力具有最高的影响。这在瞬时转染有pJTI-R4-PylRS-eGFPY40以及编码不同数目的U6-tRNA表达盒的载体的细胞中观察到,并且使用Accuri流式细胞仪确定平均荧光(图4A)。我们观察到,缺少tRNA表达盒的细胞或在不存在ALOC的情况下生长的细胞不显示显著的GFP信号。然而,GFP的表达水平随着tRNA基因拷贝的数目增加,指示tRNA是琥珀压制系统的重要组分。为了进一步细化tRNA水平在琥珀压制中的影响,我们瞬时转染不同量的编码pylRS或tRNA基因的载体,并且判定在存在2mMALOC的情况下在包含琥珀终止密码子的靶蛋白质上的琥珀压制效率。当将编码人细胞因子(在氨基酸残基131处包含琥珀密码子的FGF21)的表达构建体(此处起始子蛋氨酸是1-SEQID63,SEQID64)与pylRS以及6个基因拷贝的U6-tRNA盒共转染时,我们观察到截短体至全长FGF21的大约50%转化(图4B)。在转染中将pylRS载体的量加倍不显著改变截短和全长FGF21的比率(2xpylRS)。然而,引入另外拷贝的tRNA盒(15xU6-tRNA)导致全长FGF21的相对表达上的显著增加。这指明,tRNA水平对琥珀压制是最大的限制因子。因此,产生具有有效琥珀压制特性的细胞系需要高水平的tRNA表达。然而,诸位发明人发现,表达这些tRNA质粒特别对细胞生长有害。如与亲本系进行比较,这些tRNA表达盒的内在毒性反映在可观察到的形态变化以及高功能性平台细胞的生长速率降低。当该平台细胞系的效力直接受tRNA水平影响时,高水平的tRNApyl导致细胞毒性影响。诸位发明人观察到,虽然引入大数目的U6-tRNA基因改善在存在nnAA以及pylRS的情况下的琥珀压制,但高水平的tRNA也导致细胞毒性。为了证实此影响是否与tRNA表达有关,将针对pSB-9xtRNA(DG44-CHO-191)的存在选择的CHO细胞系,在CMV启动子(pCEP4-PylRS)控制下,用编码eGFPY40(P5-P2eGFPY40)的载体单独或与编码pylRS的载体或在包含pOriP元件(pOriP-9xtRNA)的质粒中包含九拷贝的U6-tRNA的载体组合进行瞬时转染,并允许该质粒在表达EBNA-1的细胞中长时间保留(单(Shan)2006以及EP1992698,通过引用以其全文结合在此),并将这些细胞在37C孵育48h。使用流式细胞仪(图4C)对荧光水平进行定量。虽然在表达pyltRNA的细胞的背景下添加pylR表达盒在不存在nnAA的情况下导致增加的琥珀压制水平时,但此影响在用另外拷贝的tRNA转染的细胞中扩大。这些数据表明,高水平的tRNApyl远高于野生型细胞地诱发背景琥珀压制水平。与琥珀压制相关的毒性记录于针对各种系统的文献中,并且很大程度上归因于正常地在琥珀密码子处终止的必要基因的伸展(利伯曼以及谢尔曼1976;利伯曼等人,1976)。当前的想法是,这些基因向其天然截止外伸展可改变、降低或消除这些蛋白质的功能。最终,为了确定琥珀压制是否导致抑制细胞生长作用,将1000个HEK293c18细胞铺板在96孔板上并且用pCEP4-pylRS以及pOriP-9xtRNA构建体瞬时转染。使细胞生长于包含滴定的nnAA(ALOC)(从5mM开始至0.08mM)的DMEM-C培养基上。使用MTS比色测定在转染时以及生长5天之后来测定细胞生活力(图4D)。该数据显示,甚至小浓度的nnAA也导致抑制细胞生长作用。在当前的假设中,与琥珀压制相关的毒性是该系统固有的并且不能被避免。因此,包含tRNApyl以及PrylRS的平台细胞系不适于制造基于蛋白质的药物,这需要如通过每个细胞产生的蛋白质的量测量的高产率。然而,在转染包含琥珀密码子的靶蛋白质以及随后的选择后,诸位发明人观察到,这些细胞重新获得纺锤形和扁平的外观,这是未经转染的细胞的特征并且显示改善生长速率(图4E)。这表明,高水平的包含琥珀密码子的信使的存在吸收了该背景琥珀压制并且限制了对必要基因的影响。因此,细胞系的构建可能需要包含琥珀密码子的先前存在的以及高表达的靶,使得能够分离非常高功能的琥珀压制细胞。实例3-缓和琥珀压制相关的毒性的技术与琥珀压制相关的毒性使我们想起如下可替代的途径,这些途径将缓和在开发表达细胞系中的此毒性,同时使能够分离高活性琥珀压制细胞。“靶标第一”途径缓和在开发稳定表达细胞系中观察到的毒性同时使能够分离高活性琥珀压制细胞的一个途径需要在引入pylRS以及pyltRNA之前引入高表达的包含琥珀密码子的靶基因。来自此基因的高水平的信使有力地与内源基因表达竞争在该细胞中可获得的活化pyltRNA,并且因此减小对该细胞的功能性机构的影响。为此将真核表达宿主细胞用意在表达的并且包含一个或多个琥珀终止密码子的基因转染,例如克隆到载体pOptivec(生命技术公司)中的IgG。将经转染的细胞凭借其对生长于缺少HT的培养基以及缺少HT并补充有10nM,50nM或100nMMTX的培养基的抗性和能力进行选择。通过将1-50个细胞转移至96-孔板的每个孔中并且允许殖于该孔,对存活的细胞进行克隆。然后通过ELISA对孔进行测定,以鉴别包含高滴度的截短抗体的孔。为此,将ELISA板用磷酸盐缓冲盐水(PBS)中的抗原(例如1ug3xFLAG-IL-6-Avor0.5ug/mLHer2细胞外结构域)在4℃涂覆1h或过夜。在包含10%山羊血清或1%BSA的PBS中洗涤并且阻滞之后,在室温下,向适当的孔中添加40ul的包含10%山羊血清或35ul的1%脱脂乳的PBS以及10ul的表达培养基持续1h。将孔在水中洗涤若干次,并且在室温向每个孔中添加50ul的1:4,000稀释的共轭至辣根过氧化物酶的第二抗体(抗-人类κ-HRP)(杰克森实验室)持续1h。然后洗涤各孔并将50ul的SureblueReserveTMB(KPL)添加到每个孔。5-10分钟之后通过添加0.1NH2SO4停止显色,并使用读板仪在450nM波长定量呈色。将已知浓度的对照IgG用于建立标准曲线。繁殖包含表达高水平的截短IgG的细胞的孔并扩展。将用于引入nnAA的功能性元件引入,并顺序地或同时地进行选择。在一个实例中,将显示高表达的靶基因的细胞用pMOAV2或pMOAV2puro(包含针对pylRS以及pyltRNA的基因)转染。将经转染的细胞在包含2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%胎牛血清,以及0.5mg/mL潮霉素或7.5ug/ml嘌呤霉素的DMEM中进行选择。繁殖存活的细胞并且将来自此群体的1-50个细胞接种到96孔板的每个孔中。一旦这些细胞扩张并且具有菌落形式,则将细胞暴露于2mM的nnAA,并且使用ELISA测定对其进行功能性地测定,以鉴别具有大于40%或50%或优选地大于60%或80%或90%的琥珀压制效率的克隆。为了定量全长IgG表达,在4℃,将ELISA板用磷酸盐缓冲盐水(PBS)中的1ug/mL抗-人类FC(杰克森实验室)抗体涂覆1h或过夜。在包含10%山羊血清的PBS中洗涤并且阻滞之后,在室温下,向适当的孔中添加40ul的包含10%山羊血清的PBS以及10ul的表达培养基持续1h。将孔在水中洗涤若干次,并且在室温向每个孔中添加50ul的1:10,000稀释的共轭至辣根过氧化物酶的第二抗体(抗-人类H+L-HRP)(杰克森实验室)持续1h。然后洗涤各孔并将50ul的SureblueReserveTMB(KPL)添加到每个孔。5-10分钟之后通过添加0.1NH2SO4停止显色,并使用读板仪在450nM波长定量呈色。将已知浓度的对照IgG用于建立标准曲线。这个测定将确定全长IgG的表达水平。为了确定截短的IgG的水平,将ELISA板用磷酸盐缓冲盐水(PBS)中的抗原(例如1ug/mL3xFLAG-IL-6-Avor0.5ug/mLHer2细胞外结构域)在4℃涂覆1h或过夜。在包含1%BSA的PBS中洗涤并且阻滞之后,在室温下,向适当的孔中添加35ul的包含0.1%脱脂乳的PBS以及15ul的表达培养基持续1h。将孔在水中洗涤若干次,并且在室温向每个孔中添加50ul的1:10,000稀释的共轭至辣根过氧化物酶的第二抗体(抗-人类κ-HRP)(杰克森实验室)持续1h。然后洗涤各孔并将50ul的SureblueReserveTMB(KPL)添加到每个孔。5-10分钟之后通过添加0.1NH2SO4停止显色,并使用读板仪在450nM波长定量呈色。将已知浓度的对照IgG用于建立标准曲线。确定在每个孔中的截短的与全长的IgG的比率,并且对显示高琥珀压制活性的、全长IgG水平是总共产生的IgG的至少25%或50%,优选地40%-60%或80%-90%或更高的孔进行繁殖。如果必要,则将另外的tRNA基因引入这些被选择的池的细胞中,以进一步改善该琥珀压制效率。为此,将pSB-9xtRNA-MAR表达盒转染到这些细胞中,并且凭借在包含2mMglutamax、1mM丙酮酸钠、6mM谷氨酰胺、1x非必要氨基酸(GibcoCAT#11140-050)、10%胎牛血清、以及0.5mg/mL潮霉素或7.5ug/ml嘌呤霉素以及5ug/mL杀稻瘟素(DMEM-BSD))的DMEM或可替代地包含8mM谷氨酰胺、0.5mg/mL潮霉素或7.5ug/ml嘌呤霉素以及5ug/mL杀稻瘟素(ProCHO4-BSD)的ProCHO4(隆萨(Lonza))或等效培养基中的抗生素抗性选择转染子。使用上述的ELISA测定选择具有最高活性的tRNA的细胞,以确定暴露于nnAA的细胞中的全长和截短IgG产率。对比亲本细胞显示改善的全长IgG与截短IgG比率的细胞进行繁殖。如果需要另外的tRNA基因插入的话,如上所述的用pSZ-9xtRNA重复该方法,并且在包含5ug/mL博莱霉素的培养基中选择细胞,随后进行功能性选择筛选。“诱饵蛋白质”途径缓和在开发稳定表达细胞系中观察到的毒性同时使能够分离高活性琥珀压制细胞的替代途径涉及引入包含琥珀密码子的以高水平表达的替代基因,其中在靶基因的表达过程中,表达是通过诱导型启动子驱动以使得能够下调其表达。这具有如下优点,可产生表达PylRS/tRNApyl正交机构的稳定细胞系,并且用于修饰多个靶。为此,将真核表达宿主细胞(例如CHO细胞)用意在用于表达的并且包含一个或多个琥珀终止密码子的基因进行转染,例如但不限于GFP、eGFP、红色荧光蛋白、谷胱甘肽–S-转移酶、b-微球蛋白、或B-半乳糖苷,克隆到优选地包含诱导型启动子,例如Tet-On3G(Clonthech)、T-Rex(生命技术公司)、蜕皮激素-可诱导型、或类固醇-诱导型启动子的哺乳动物表达载体中。将经转染的细胞凭借其对生长于包含适当的抗生素的培养基中的抗性以及能力进行选择。通过将1-50个细胞转移至96-孔板的每个孔中并且允许殖于该孔,对存活的细胞进行克隆。然后通过ELISA测定对孔进行测定,以鉴别包含高滴度的截短蛋白质的孔。包含一个或多个琥珀密码子的高表达的替代蛋白质将起琥珀槽的作用,以吸收琥珀压制活性并保护该细胞免受琥珀压制的有害影响。将用于引入nnAA的功能性元件(例如U6-tRNA盒以及pylRS基因)引入宿主细胞,并顺序地或同时地进行选择。在一个实例中,将显示高表达的替代靶基因的细胞用pMOAV2或pMOAV2puro(包含针对pylRS以及pyltRNA的基因)转染。将经转染的细胞在包含2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%胎牛血清,以及0.5mg/mL潮霉素或7.5ug/ml嘌呤霉素的DMEM中进行选择。繁殖存活的细胞并且将来自此群体的1-50个细胞接种到96孔板的每个孔中。一旦这些细胞扩张并且具有菌落形式,则将细胞暴露于2mM的nnAA,并且使用ELISA测定对其进行功能性地测定,以使用报告蛋白(eGFPY40)或该替代靶蛋白质鉴别具有大于40%或50%或优选地40%-60%或大于80%或90%的琥珀压制效率的克隆。通过限制稀释克隆或细胞分选分离高功能克隆。然后将编码蛋白治疗剂的基因引入这些细胞并进行选择。分离高表达克隆并且通过ELISA测定鉴别。然后将高表达克隆针对琥珀压制效力进行筛选。确定在每个孔中的截短与全长IgG的比率,并且对显示高琥珀压制活性的孔、显示包含nnAA的全长蛋白质的至少40%或50%但优选地40%-60%或80%-90%或更大的琥珀压制水平的克隆进行繁殖。如在上文中所讨论的,如果必要,则将另外的tRNA基因引入这些被选择的池的细胞中,以进一步改善该琥珀压制效率。“阻抑型tRNA”途径设计一种可替代的具有调节tRNA表达水平以及使得tRNA相关的细胞毒性减轻策略。为此,对tRNA表达所必须的启动子元件(例如U6或H1)进行修饰以包括使基因表达抑制的序列元件(如TetO阻抑物元件)。这使得在生长阶段tRNA的表达下调以及在靶基因表达阶段tRNA表达的诱导。“诱饵氨基酸”途径已通过引入氨基酸类似物工程化了调节背景琥珀压制的影响的可替代策略,该氨基酸类似物由pylRS识别,并活化为tRNApyl,但被修饰以不允许肽键形成。将此诱饵氨基酸活化到tRNApyl上会与通过宿主RSs或pylRS进行的天然氨基酸活化有力地竞争,并且产生诱饵氨基酸活化的tRNA的细胞池。此池也将与错误酰化的(mis-acylated)tRNApyl竞争琥珀密码子。因此该池的诱饵氨基酸的活化tRNA将允许蛋白质合成在琥珀终止密码子处的正常终止。在平台细胞系构建的过程中,细胞例如DG44CHO细胞生长于包含诱饵氨基酸的培养基中,并且将编码tRNA以及pylRS的基因稳定整合到这些细胞中。将经转染的细胞通过生长于包含适当的抗生素的培养基中进行选择,并且扩展存活的细胞。将此池用编码eGFPY40的载体进行瞬时转染,并且使细胞生长于包含允许肽键形成使得能够通读琥珀密码子的nnAA并且缺少诱饵氨基酸的培养基中。然后凭借eGFPY40报告基因的表达水平鉴别高功能细胞,并且使用流式细胞术使用BDFACSAriaII分离细胞。将经分选的细胞扩展,并且使用可获得的报告基因(例如eGFPY40或FGF21-131amb)判定在这个经分选的池中的琥珀压制效率。如果必要,可进行tRNA或pylRS基因的反复添加以及使用流式细胞术进行的选择,以增强该琥珀压制的效率。然后将平台细胞用在包含DHFR基因的载体例如Optivec质粒(生命技术公司)或包含Glut胺合成酶基因(隆萨)的质粒中的靶基因例如IgG(针对所希望的抗原,包含琥珀密码子)进行转染,以允许基因表达选择。使表达高水平的截短蛋白的细胞生长在适当的选择(分别是甲氨蝶呤或氨基亚砜蛋氨酸)下,以针对高表达细胞进行选择。将克隆使用限制细胞稀释进行分离,并且使用ELISA测定鉴别能够具有有效的琥珀压制以及高表达产率的细胞。实例4:修饰靶蛋白质使nnAA能够掺入在载体pTracerHisEF/HISA(生命技术公司)中融合的绿色荧光蛋白质-杀稻瘟菌素的ORF中引入琥珀密码子以产生GFPY40报告子构建体。简单地说,使用定点诱变改变该GFPORF在位置+120处(其中+1是该起始密码子的A)的单核苷酸,将胞嘧啶变为鸟嘌呤,并由此产生框琥珀终止密码子。使用重叠寡聚物以及PCR通过基因合成产生FGF21ORF以再生人类FGF21序列,如在SEQID61核苷酸序列中所示;SEQID62氨基酸序列),包含另外的氨基末端3xFlag标签(编码dykdhdgdykdhdidykddddks)(3xFLAG-FGF21,SEQID80)。克隆构建体到pJ201穿梭载体中并且通过使用HinDIII以及XhoI限制酶消化转移以及连接到pCEP4(生命技术公司)。产生的构建体将FGF21的ORF置于下游并且在用于哺乳动物细胞表达的CMV启动子控制下。通过在该FGF21ORF的F12(SEQID66)、L66(SEQID68)、P90(SEQID70)、R131(SEQID64)、以及P140(SEQID71)位置进行定点诱变引入琥珀密码子。使用两步PCR扩增方案,使用重叠寡聚物将3xFLAG标签取代为6xHis标签。简单地说,设置两个PCR反应,一个扩增CMV启动子而第二个扩增FGF21ORF且框架内有5’6xHis标签。然后在第三个PCR反应中使用侧翼寡聚物以将CMV启动子加入到6xHis-FGF21构建体。通过Gateway将产物克隆进入pDONR221P4r-P3r载体中以产生6xHIS-FGF21wt和6xHIS-FGF21R131两者。修饰针对IL-6的抗体以使得能够进行nnAA的整合及其随后的共轭。为了产生这种分子,通过PCR并且克隆进pFUSE-CHIg-hG1(重链)和pFUSE-CHLIg-hK(轻链)载体(英杰公司)中将针对人细胞因子IL-6的兔抗体的可变区移植到人框架上(参见WO12032181)以产生兔-人杂交体。也邻近IL-6CDR掺入另外的突变以人源化该抗体。所得到的载体对pFuse-28D2γ和pFUSE-28D2κ通过瞬时转染用于抗-IL-6IgG的共转染和表达。通过定点诱变和经由测序的突变体筛选在所希望的位点引入琥珀密码子而产生nnAA掺入位点。此导致在位点274处包含琥珀密码子的重链克隆(pFuse-28D2γ_K274am)(SEQID41)。重链构建体和轻链构建体的共转染允许抗-IL-6抗体表达。通过TOPO克隆将包含抗-IL-6IgG重链(在位置K274处包含琥珀)的整合构建体克隆到pOptivec中。通过PCR扩增轻链表达构建体(包括其启动子和多聚A序列)并且将tRNA的单拷贝通过两步骤PCR方法使用重叠寡聚物进行连接,并克隆到可用位点到包含重链的pOptivec质粒中(pOtivec-28D2-GKt)。执行这些抗体的瞬时表达和稳定表达以整合赖氨酸-叠氮化物、ALOC、炔丙基-赖氨酸以及赖氨酸-氯化物nnAAs。表达以及纯化所有的实验蛋白质从稳定细胞系分离(实例1)。可替代地,利用瞬时转染细胞系,将CHO或HEK293细胞铺板至大约90%汇合且在37C生长。翌日,根据具体的制造商说明,将这些铺板的细胞与适当的DNA进行孵育,该DNA之前用亲脂性试剂(脂质体转染(Lipofectamine)2000,293fectin(英杰公司(invitrogen))处理过。在存在nnAA、ALOC、Lys-叠氮化物、炔丙基赖氨酸或Lys-氯化物)的情况下生长2-5天后,收获该生长培养基,并且直接使用,或将所表达的蛋白质通过适当的方法纯化。为了IgG的表达,使细胞生长于包含低IgG胎牛血清的培养基中。使经稳定转染的细胞系附着地生长于烧瓶中,至90%汇合,并暴露于nnAA(选自以下:ALOC(N-烯丙氧基羰基-L-赖氨酸)、lys叠氮化物、炔丙基赖氨酸、(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸(式VI.1))持续5-7天,并且收获该生长培养基,并且直接使用,或将所表达的蛋白质通过适当的方法纯化。为了IgG的表达,使细胞生长于包含低IgG胎牛血清的培养基中。在真核细胞的稳定或瞬时表达之后,从生长培养基纯化此处所述的表达的IgGs、scFvs、或FGF21。在每种情况下,将0.1体积的10xPBS添加到表达上清液,以平衡这些盐以及该样本的pH。为了纯化6xHis标记的蛋白质,将上清液在4℃用PBS透析持续16。通过批结合或重力流使蛋白质结合到镍-NTA珠,并且用洗涤缓冲液(配方)充分洗涤。将结合的材料进行洗脱(50mM磷酸钠pH7.4、300mMNaCl、250-500mM咪唑)。通过SDS-PAGE以及考马斯亮蓝染色鉴别包含靶蛋白质的级分。汇集峰级分,并且在进一步使用之前用PBS进行透析。通过蛋白质A亲和色谱法对IgG进行纯化。简而言之,将表达上清液补充以0.1体积的10xPBS,并且穿过1mL或5mL蛋白质A琼脂糖凝胶快流柱(GE)。将结合的材料用5-10柱体积的PBS洗涤并且用3-5体积的0.1M甘氨酸pH3.0进行洗脱。随后通过添加0.05体积的20xPBS中和级分,以达到中性pH。将洗脱级分通过SDS-PAGE以及考马斯亮蓝染色进行分析,并且汇集峰蛋白质级分,并且在4oC用PBS透析持续16小时。使用此方法进行制备:修饰抗-IL-6-Lys叠氮化物274h,FGF21,以在位置131包括NNAA(S)-2-氨基-6((丙-2-炔基氧基)羰基氨基)己酸(Lys-炔烃)实例5:包含nnAA的蛋白质的共轭在重链的位置274掺入NNAALys-叠氮化物的抗-IL-6抗体(抗-IL-6-Lys叠氮化物274h)与20KPEG末端炔烃的PEG化在具有小的磁力搅拌器的8x30mm玻璃小瓶中放置TBTA(80mM,3.75mL)的二氯甲烷溶液,通过在该管上温和地吹氮气蒸发溶剂。向其中添加磷酸盐缓冲液(125mM,pH=7.4,53uL)以及20KPEG炔烃(3mM,33uL)的水性溶液。添加抗-IL-6-Lys叠氮化物274h(0.4mg/mL,6.26uL)的溶液,随后添加半胱氨酸(100mM,2uL)以及硫酸铜(80mM,1.9uL)的溶液。将该小瓶用氩气覆盖,加盖并且温和混合4h。将该反应混合物的一部分去除(15uL)并且与非还原胶装载缓冲液(4X,NuPage,英杰公司,7.5uL)混合。将整个体积装载到SDS-PAGE凝胶上用于分析(图5A):SDS-PAGE指示,这些铜条件产生单PEG化以及双-PEG化抗体种类的混合物。PDSi密度测定法指示,单PEG化种类处于大约1:1比率(单=47%,双=53%)。不具有叠氮化物的抗体不能在相似的条件下发生反应,证明对该叠氮化物的特异性。在重链的位置274掺入NNAALys-叠氮化物的抗-IL-6抗体(抗-IL-6-Lys叠氮化物274h)与20KPEG环辛炔(双环[6.1.0]壬-4-炔-连接的PEG)的PEG化在具有小的磁力搅拌器的8x30mm玻璃小瓶中放置磷酸盐缓冲液(125mM,pH=7.4,60uL)以及20KPEG环辛炔(双环壬炔)(3mM,33uL)的水性溶液。添加抗-IL-6-Lys叠氮化物274h(0.4mg/mL,6.26uL)的溶液,并将该小瓶加盖,并且温和混合4h。将该反应混合物的一部分去除(15uL)并且与非还原胶装载缓冲液(4X,NuPage,英杰公司,7.5uL)混合。将整个体积装载到SDS-PAGE凝胶上进行分析。SDS-PAGE凝胶分析(图5B:泳道1:用20KPEG环辛炔处理的(抗-IL-6-Lys叠氮化物274h),泳道2:用20KPEG环辛炔处理不具有叠氮化物的抗体,泳道3:未经处理的抗体),指示单PEG化以及双-PEG化抗体种类的混合物。所得到的凝胶的密度测定指示单PEG化(10%)以及双-PEG化抗体(76%)的混合物,起始材料被用光。包含叠氮化物的抗体是唯一发生反应的种类。为了测试针对IL-6的抗体的活性,并且确定此抗体的修饰是否改变了此抗体的结合特性,我们建立了体外IL-6中和测定。为此,将IL-6依赖性鼠B-细胞杂交瘤细胞(B9)接种到96孔板中在包含50pg/mLIL-6的培养基中。向一系列的孔中添加不同浓度的抗-IL-6抗体以及对照,并且在37oC生长3天。然后使用阿尔玛蓝确定这些细胞的生活力,阿尔玛蓝是允许定量地测量细胞的健康的比色测定。简而言之,向每个孔中添加25uL的试剂,并且允许细胞继续生长8-16小时。在孵育之后,在570nm以及600nm波长用分光光度计读出这些组。绘制测量的吸光度比对相应的抗体浓度的图。该数据指示,与裸的未经修饰的抗体进行比较,抗-IL-6抗体的位点特异性PEG化未降低该抗体的功能性特征。IL-6抑制测定(图8C)。为了测试针对IL-6的抗体的活性,使用IL-6中和测定。将IL-6依赖性B9细胞接种到96孔板中在包含50pg/mLIL-6的培养基中。向一系列的孔中添加不同浓度的抗-IL-6抗体以及对照,并且在37℃生长3天。使用阿尔玛蓝确定这些细胞的生活力。简而言之,向每个孔中添加25uL的试剂,并且允许细胞继续生长8-16小时。在孵育之后,在570nm以及600nm波长用分光光度计读出这些组。绘制测量的吸光度比对相应的抗体浓度的图。制备抗-IL-6(抗体)-抗IL-23(scFv-PEG)双特异性针对人细胞因子IL23的scFv的产生,这是使用使能够位点特异性地在所希望的位点掺入叠氮基-高丙氨酸的大肠杆菌表达系统产生的。简而言之,将大肠杆菌(B834)的蛋氨酸营养缺陷型菌株用编码scFv的表达质粒转化为hIL23(WO2012/032181,通过引用结合在此)。scFv基因仅编码两个蛋氨酸,翻译后裂解的起始蛋氨酸以及在该分子的c-末端的蛋氨酸。将这些转化的细胞在丰富培养基中发酵,并且该培养允许生长达到生长平台期。在此时,将这些细胞用IPTG诱导以对scFv的表达去阻抑,并且将该培养基补充以AHA。然后允许这些细胞生长另外四小时,并且通过离心收获这些细菌细胞。将表达的scFv从内含体中纯化并且体外折叠。表达的scFv包含叠氮化物,该叠氮化物包含如下氨基酸,该氨基酸允许与包含炔烃的部分发生共轭。为了产生双特异性抗体构建体,将抗-IL-6-Lys叠氮化物274h用双-炔烃PEG部分连接到抗-IL23scFv(WO2012/032181,通过引用接合在此)。为此,在具有小的磁力搅拌器的8x30mm玻璃小瓶中放置磷酸盐缓冲液(20mM,pH=7.4,80uL)。添加抗-IL-6-Lys叠氮化物274h的溶液(0.4mg/mL,6.3uL),随后添加之前共轭至-20KPEG炔烃的抗-IL23scFv的溶液(1mg/mL,2.1uL)。向此溶液中添加BME的溶液(100mM,3uL)以及硫酸铜的溶液(80mM,2.81uL)。允许将该混合物搅拌4h。将该反应混合物的一部分去除(15uL)并且与非还原胶装载缓冲液(4X,NuPage,英杰公司,7.5uL)混合。将该整个体积装载到SDS-PAGE凝胶上进行分析。产生了非常小量的双特异性物,如通过抗体带上面的新的更高的分子带证明的(图6A)。关于还原胶,将该反应混合物的一部分去除(15uL)并且与还原胶装载缓冲液(4X,NuPage,英杰公司,30%BME,7.5uL)混合。将该整个体积装载到SDS-PAGE凝胶上进行分析。产生了非常小量的双特异性物,如通过重链带上面的新的更高的分子带证明的,与预期MW变化一致(图6B)。FGF-21(被修饰以包括NNAALys-炔烃)与20K线性PEG双叠氮化物的PEG化对于所有实验,使用了标准转染条件。简而言之,将之前针对稳定的pylR表达选择的293个细胞的克隆铺板至大约90%汇合,并且在37oC生长16h。翌日,根据具体制造商说明,将这些铺板的细胞用之前与亲脂性试剂(脂质体转染2000,293fectin(英杰公司)组合的6xHIS-FGF21R131进行处理。然后使细胞生长于包含2mM炔丙基-赖氨酸nnAA的DMEM完全(DMEM,2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸,10%胎牛血清)培养基上持续5-7天。收获该生长培养基,并且将FGF21通过亲和色谱法在5mL预包装的镍-NTA柱(GE)上进行纯化。将表达的6xHIS-FGF21从该生长培养基中纯化。这里,将0.1体积的10xPBS添加到表达上清液,以平衡这些盐以及该样本的pH。并且在4oC将该培养基用PBS透析16。通过批结合或重力流使表达的FGF21结合到镍-NTA珠(GE)上,并且用洗涤缓冲液(50mM磷酸钠pH7.4,300mMNaCl,20mM咪唑)充分洗涤。将结合的材料用NiNTA洗脱缓冲液(50mM磷酸钠pH7.4、300mMNaCl、250-500mM咪唑)进行洗脱。通过SDS-PAGE以及考马斯亮蓝染色鉴别包含靶蛋白质的级分。汇集峰级分,并且在进一步使用之前用PBS进行透析。在具有磁力搅拌器的20mL小瓶中放置被修饰以在位置131(SEQID64)包括NNAA(S)-2-氨基-6((丙-2-炔基氧基)羰基氨基)己酸(Lys-炔烃)的FGF21的溶液(20ug/mL,0.001mM,5000uL)。向其中添加20K线性PEG双-叠氮化物的溶液(60mg/mL,1.67mL)。添加SDS的溶液(20%,250uL)以及DTT的溶液(250mM,60uL)。TBTA的DMSO溶液(80mM,7.96uL)以及硫酸铜的水性溶液(80mM,94uL)。将该小瓶加盖,并允许将该反应搅拌过夜。将该混合物离心(10000g,15min)并保留上清液。将该反应混合物通过SDS-PAGE进行评估,并且观察到明显的分子量位移,与20kDaPEG至该多肽的共轭一致(图7A)。为了评估该PEG化FGF21构建体的效价,将肥胖的db/db小鼠每天用0.25mg/KgFGF21或20KPEG-FGF21进行处理,并且在处理三次之后使用手持型葡萄糖监测仪测量饲养的小鼠中的葡萄糖水平(图7B)。纯合的db/db小鼠(B6.BKS(D)-Leprdb/j,杰克森实验室)是针对糖尿病的良好表征的动物模型,并且在3-4周龄之后变得肥胖。这些动物也显示升高的血浆胰岛素、血糖、以及延迟的伤口愈合。在此研究中,向7-8周大的雄性db/db小鼠随意喂食实验室饮食(LabDiet)5053鼠食物20(RodentDiet20)。使小鼠适应七天,并且随后每天皮下给予PEG化FGF21、未经修饰的FGF21以及运载体(PBS)持续十一天。每天向每个小鼠给予0.25mg/Kg的PEG-FGF21或未经修饰的FGF21持续三天。在第3天给予化合物一小时之后,通过夹尾出血确定喂食的血糖水平,并且通过手持型葡萄糖仪表(拜耳(Bayer))测量葡萄糖水平。该数据显示,在氨基酸残基131处PEG化的FGF21具有与野生型FGF21相同的效价,并且与安慰剂对照进行比较显示改善的葡萄糖水平维持。实例6:制备氨基酸以及诱饵氨基酸从商业供应商购买2-{[(苄氧基)羰基]氨基}-6-{[(丙-2-烯-1-基氧基)羰基]氨基}己酸(式VIIA.4,巴亨公司(BaChem))、2-{[(9H-芴-9-基甲氧基)羰基]氨基}-6-{[(丙-2-烯-1-基氧基)羰基]氨基}己酸(式VIIA.5,巴亨公司)、6-{[(叔丁氧基)羰基]氨基}己酸(式VIIB.4)。制备具有式VIIA的诱饵nnAA通过用活化亲电子试剂酰化起始氨基酸的ɑ-氨基基团容易地制备式VII类似物。这是通过将该起始材料用酰基氯、活化酯、酸酐或磺酰氯处理进行的。然后可利用该产物用于细胞系开发。制备具有式VIIB的诱饵氨基酸:制备6-{[(丙-2-烯-1-基氧基)羰基]氨基}己酸(式VIIB.1)。在具有磁力搅拌器的20mL小瓶中放置6-氨基己酸(280mg)、氢氧化钠(1M,5.3mL)以及二噁烷(2mL)。添加烯丙基氯甲酸酯(228uL)并且将混合物搅拌3h。将该混合物用1M柠檬酸处理直至pH是酸性。将该混合物用乙酸乙酯(x3)萃取并且保留有机层。合并有机层,用硫酸钠干燥,过滤并浓缩。分析MS:m/z(ES+)计算215.1(M+H)+,观察到216.1。制备6-{[(2-氯乙氧基)羰基]氨基}己酸(式VIIB.3)。在具有磁力搅拌器的20mL小瓶中放置6-氨基己酸(1180mg)、氢氧化钠(1M,22.5mL)以及二噁烷(23mL)。添加2-氯乙基氯甲酸酯(932uL)并且将混合物搅拌3h。将该混合物用过量的1M柠檬酸处理直至pH是酸性。将该混合物用乙酸乙酯萃取(x3)并且保留有机层。合并有机层,用硫酸钠干燥,过滤并浓缩。分析MS:m/z(ES+)计算237.1(M+H)+,观察到238.1。制备6-{[(2-叠氮基乙氧基)羰基]氨基}己酸(式VIIB.6)。在具有磁力搅拌器的20mL小瓶中放置6-{[(2-氯乙氧基)羰基]氨基}己酸(250mg)以及DMSO(5mL)。添加叠氮化钠,并将2-氯乙基氯甲酸酯(70mg)加热至60℃并且搅拌20h。将该混合物用水(5mL)稀释并且倾倒至1M柠檬酸(10mL)上。将该混合物用乙酸乙酯萃取(x3)并且保留有机层。合并有机层,并且用5%氯化锂洗涤。将有机层用硫酸钠干燥,过滤并浓缩。分析MS:m/z(ES+)计算244.1(M+H)+,观察到245.2。制备6-{[(丙-2-炔-1-基氧基)羰基]氨基}己酸(式VIIB.5)。在具有磁力搅拌器的20mL小瓶中放置6-氨基己酸(220mg)、氢氧化钠(1M,4.2mL)以及二噁烷(2mL)。添加炔丙基氯甲酸酯(163uL)并且将混合物搅拌3h。将该混合物用过量的1M柠檬酸处理直至pH是酸性。将该混合物用乙酸乙酯萃取(x3)并且保留有机层。合并有机层,用硫酸钠干燥,过滤并浓缩。分析MS:m/z(ES+)计算213.1(M+H)+,观察到214.1。制备5-{[(丙-2-烯-1-基氧基)羰基]氨基}戊酸(式VIIB.2)。在具有磁力搅拌器的500mL圆底烧瓶中放置5-氨基戊酸(15.0g)、水(100mL)以及2N碳酸钠(40mL)。逐滴添加于二噁烷(100mL)中的烯丙基氯甲酸酯(8.2mL),并且将该最终混合物搅拌3h。将该混合物用2NHCl(~50mL)进行酸化。将该混合物用乙酸乙酯(4x100mL)萃取并且保留有机层。合并有机层,用硫酸钠干燥,过滤并浓缩。分析MS:m/z(ES+)计算201.1(M+H)+,观察到202.1。实例7.制备式V和VI类似物制备(S)-2-氨基-6((2-氧代-2-苯基乙酰胺)己酸(式V.6)在具有磁力搅拌器的50mL圆底烧瓶中,将丙酮酸(3.5g,23.3mmol)溶解在二氯甲烷和DMF(20mL)的2:1混合物中。向该混合物中添加DCC(5.7g,27.6mmol)和NHS(3.2g,27.6mmol)。伴随着搅拌,将该混合物加热至50℃持续30min。允许将该溶液冷却,并且然后通过过滤器添加到在单独的具有磁力搅拌器的100mL圆底烧瓶中的N-Boc-赖氨酸(5.2g,21.2mmol)于DMF(20mL)中的悬浮液中。在添加活化酯之后,添加三乙胺(8.8mL,63.6mmol),并将该混合物搅拌过夜。将该混合物在乙酸乙酯与柠檬酸之间分配。将各层分离并且将水层用乙酸乙酯萃取4次。合并有机层,用硫酸钠干燥并浓缩。将所得到的残余物进一步通过快速色谱法纯化,以给予最终的呈油状的N-Boc赖氨酸衍生物。在100mL圆底烧瓶中放置乙腈(50mL)中的酮基-N-Boc赖氨酸衍生物(4g,10.6mmol)。向其中添加盐酸(15mL,4N于二噁烷中)的溶液。将该溶液搅拌2h并且浓缩。最终通过快速色谱法纯化,给予靶氨基酸。分析MS:m/z(ES+)计算278.1(M+H)+,观察到279.1。制备(2S)-2-氨基-6-(2-叠氮基乙酰胺基)己酸(式V.8)在25mL圆底烧瓶中放置悬浮于二噁烷(5mL)中的N-Boc-赖氨酸(500mg,2.0mmol)。添加饱和NaHCO3(2mL)并将该溶液冷却至0℃。缓慢添加于二噁烷(2mL)中的溴乙酰氯(169uL,2.0mmol)。允许将该溶液在0℃搅拌1h并且然后在室温4h。将该溶液转移到萃取漏斗并在水和醚之间分配。将有机层去除并用柠檬酸使水层成为酸性(pH=2)。用乙酸乙酯(3x50mL)萃取水层,合并有机层并用硫酸钠干燥,过滤并且浓缩。将所得到的残余物转到下一步。在50mL圆底烧瓶中放置粗的于二噁烷(10mL)中的N-Boc--2-溴乙酰基-赖氨酸(740mg,2.0mmol)。向其中添加叠氮化钠(10mL,1M)的溶液。将该溶液在60℃下搅拌过夜。将该混合物在柠檬酸(1M,50mL)和乙酸乙酯(100mL)之间分配。保留有机层,并且将水层萃取另外3次。合并有机层,用硫酸钠干燥并浓缩为油。将粗的N-Boc--2-叠氮基-乙酰基-赖氨酸溶解于乙腈(10mL)中并添加TFA(2mL)。将该混合物搅拌2h,并且然后浓缩。将该溶液用甲苯(10mL)处理并浓缩(2X),并且用乙腈(10mL)处理并浓缩(2X)。将残余物在真空下干燥过夜。将残余物吸收于MeOH中并用甲基-叔丁醚沉淀。将粘性油通过离心分离,将上清液进行处置。分析MS:m/z(ES+)计算229.1(M+H)+,观察到230.1。制备(2S)-2-氨基-6-(戊-4-烯酰胺基)己酸(式V.5)在25mL圆底烧瓶中放置悬浮于二噁烷(10mL)中的N-Boc-赖氨酸(500mg,2.0mmol)。添加1MK2CO3(5mL)并将该溶液冷却至0℃。缓慢添加于二噁烷(2mL)中的4-戊烯酰氯(224uL,2.0mmol)。允许将该溶液在0℃搅拌1h并且然后在室温4h。将该溶液转移到萃取漏斗并在水和醚之间分配。将有机层去除并用柠檬酸使水层成为酸性(pH=2)。用乙酸乙酯(3x50mL)萃取水层,合并有机层并用硫酸钠干燥,过滤并且浓缩。将所得到的残余物转到下一步。将粗的N-Boc--N-4-戊烯酰胺-赖氨酸放置在具有乙腈(5mL)和TFA(2mL)的50mL圆底烧瓶中并磁力搅拌2h。浓缩该混合物。将该溶液用甲苯(10mL)处理并浓缩(2X),并且用乙腈(10mL)处理并浓缩(2X)。将残余物在真空下干燥过夜。将残余物吸收于MeOH中并用甲基-叔丁醚沉淀。将粘性油通过离心分离,将上清液进行处置。分析MS:m/z(ES+)计算229.2(M+H)+,观察到229.1。制备羟基-正亮氨酸衍生物(式VI.1和式VI.2)伴随着磁力搅拌在100mL圆底烧瓶中放置N-Boc-羟基正亮氨酸(1g,4.1mmol)和乙腈(50mL)。将该混合物冷却到0℃并添加对硝基苯基氯甲酸酯(979mg,4.9mmol)和吡啶(2mL),并将该混合物搅拌过夜。浓缩该混合物并通过快速色谱纯化。(二氧化硅,DCM/MeOH梯度)。伴随着磁力搅拌在100mL圆底烧瓶中放置于25mL二噁烷中的2-N-Boc-乙基溴化物(1g,4.4mmol)。向其中添加叠氮化钠(1M,22.2mmol)的溶液。将该溶液在60℃下搅拌过夜。将该混合物在水与乙酸乙酯之间分配。保留乙酸乙酯层并将将水层用乙酸乙酯萃取另外三次。合并有机层,用硫酸钠干燥并浓缩为油。将油吸收于乙腈(35mL)中并添加于二噁烷中的HCL(4M,10mL)。将该混合物搅拌两小时,并在真空下浓缩。制备(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸(式VI.1)在50mL圆底烧瓶中放置于二噁烷(10mL)中的N-Boc-正亮氨酸对硝基苯基碳酸酯(503mg,1.2mmoL)。向其中添加氨基-叠氮化物(105mg,1.2mmol)于二噁烷(5mL)和吡啶(1mL)中的溶液。将该溶液搅拌过夜。将该混合物在乙酸乙酯和500mM柠檬酸之间分配。保留乙酸乙酯层并将将水层用乙酸乙酯萃取另外三次。合并有机层,用硫酸钠干燥并浓缩为油。将油进一步通过快速色谱法纯化。将分离的Boc-保护的氨基酸吸收于乙腈(15mL)中并用二噁烷中的HCl(4M,5mL)处理。将该混合物搅拌两小时,并在真空下浓缩。制备(2S)-2-氨基-6-{[(丙-2-炔-1-基)氨基甲酰基]氧基}己酸(式VI.2)在50mL圆底烧瓶中放置于二噁烷(10mL)中的N-Boc-正亮氨酸对硝基苯基碳酸酯(337mg,0.8mmoL)。向其中添加氨基-叠氮化物(135mg,2.4mmol)于二噁烷(5mL)中的溶液。将该溶液搅拌过夜。将该混合物在乙酸乙酯和500mM柠檬酸之间分配。保留乙酸乙酯层并将将水层用乙酸乙酯萃取另外三次。合并有机层,用硫酸钠干燥并浓缩为油。将油进一步通过快速色谱法纯化。将分离的Boc-保护的氨基酸吸收于乙腈(15mL)中并用二噁烷中的HCl(4M,5mL)处理。将该混合物搅拌两小时,并在真空下浓缩。实例8:抗-Her2-毒素共轭如下获得该抗-Her2抗体。针对Her2的细胞外结构域的小鼠抗体4D5的可变区是使用重叠寡聚物通过基因合成产生的并被克隆到穿梭载体中。然后这些可变区被接枝到由pFUSE-CHIg-hG1以及pFUSE-CHLIg-hK(英杰公司)编码的人框架以产生小鼠-人类杂交体。通过定点诱变,在位置274以及359(分别是SEQID47以及SEQID49)处向重链(γ)以及在位置70以及81(分别是SEQID53以及SEQID55)处向轻链(κ)中引入琥珀密码子。通过DNA测序鉴别包含该琥珀密码子的克隆。为了产生针对此IgG的在pOptivec中的整合构建体,将针对该重链的启动子和ORF通过PCR扩增,并通过限制酶消化并连接到pOptivec中而克隆。将该轻链和tRNA的单拷贝通过两步骤PCR方法使用重叠寡聚物进行连接,并克隆到可用位点到包含该重链的pOptivec质粒中。表达以及纯化通过赫塞汀CDR的基因合成产生针对HER2的抗体,并且修饰IgG1框架以使得能够进行在一个或两个位点处的nnAA的整合及其随后的共轭。将赫塞汀鼠CDR克隆到pFUSE-CHIg-hG1(重链)以及pFUSE-CHLIg-hK(轻链)(英杰公司)中以产生人源化抗体。将所得到的载体对pFuse-4D5γ以及pFUSE-4D5κ用于野生型抗-Her2IgG通过瞬时转染的共转染和表达(SEQID45,SEQID46,重链;SEQID51,SEQID52,轻链)。通过定点诱变和经由测序的突变体筛选在所希望的位点引入琥珀密码子而产生nnAA掺入位点。此导致在位置274处包含琥珀密码子的重链克隆(pFuse-4D5γ_K274am)(SEQID47)。也在pFUSE-4D5κ中构建琥珀位点。首先将终止密码子从琥珀密码子替换到赭石终止密码子,以产生载体pFUSE-4D5κ_TAA。通过定点诱变在位置D70处引入琥珀密码子(SEQID53)。通过配对这些不同的载体,可产生包含单个nnAA或两个nnAA的抗体。包含nnAA的靶抗体的瞬时表达是在稳定表达pylRS的HEK293细胞中进行的。这个细胞系是通过转染包含pCEP4(生命技术公司)中的pylRS基因的载体并且通过生长于包含潮霉素B(DMEM(生命技术公司),2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%胎牛血清,以及0.2mg/mL潮霉素)的培养基进行选择产生的。通过限制稀释克隆存活的细胞,并且扩展表明pylRS的高功能活性的克隆。这是通过在存在ALOCnnAA的情况下用编码tRNApyl的载体以及在位置Y40包含琥珀密码子的报告子构建体GFPY40瞬时转染不同克隆来实现的。使用Accuri流式细胞仪在这些细胞中定量荧光水平并且分离高功能性克隆。该抗-Her2抗体的表达是使用标准转染条件进行的。将细胞铺板至大约90%汇汇合且在37℃生长。翌日,根据具体制造商说明,将这些铺板的细胞与适当的DNA进行孵育,该DNA之前用亲脂性试剂(脂质体转染(Lipofectamine)2000,293fectin(英杰公司)处理过。在存在1-2mMLys-叠氮化物的情况下生长2-5天后,收获生长培养基,并且直接使用,或将所表达的蛋白质通过适当的方法纯化。为了IgG的表达,使细胞生长于包含低IgG胎牛血清的培养基中。在每种情况下,将0.1体积的10xPBS添加到表达上清液,以平衡这些盐以及该样本的pH,并且将抗体通过蛋白质A亲和色谱法纯化。简而言之,使表达上清液穿过1mL或5mLn蛋白质A琼脂糖凝胶快流柱(GE)。将结合的材料用5-10柱体积的PBS洗涤并且用3-5体积的0.1M甘氨酸pH3.0进行洗脱。随后通过添加0.05体积的20xPBS中和级分,以达到中性pH。将洗脱级分通过SDS-PAGE以及考马斯亮蓝染色进行分析,并且汇集峰蛋白质级分,并且在4oC用PBS透析持续16小时。包含lys叠氮化物作为nnAA的IgG被称为AzAb。制备细胞毒素-炔烃衍生物制备MMAF-炔烃衍生物。将单甲基阿里他汀F(MMAF)(6mg,8.2umol)置于小瓶中,并且添加DMSO(450uL)。将BCN碳酸酯于DMSO中的溶液(82.6ug/uL,84uL,2.4mg,8.2umol)添加到该MMAF溶液中。添加三乙胺(2.5uL,18umol),将该小瓶加盖,并且将该反应搅拌4h。分析MS:m/z(ES+)计算907.1(M+H)+,观察到908.6。制备MMAF-缬氨酸-瓜氨酸-p-氨基-苯甲酰基-碳酸酯(VCP)-环辛炔衍生物。在具有磁力搅拌器的4mL小瓶中放置MMAF(5mg,6.84umol)以及二肽val-cit-PABC-Fmoc(5.24mg,6.84umol)。向此混合物中添加DMSO(350uL)。添加乙基(羟基亚氨基)氰基乙酸酯(40mg/mL,25uL)的DMSO溶液以及叔丁醇钾(60mg/mL,25uL)的水性溶液,将该小瓶加盖并且允许搅拌过夜。分析MS:m/z(ES+)计算1358(M+H)+,观察到1359.8。将该粗混合物直接用于下一个步骤。将粗的MMAF-VCP-Fmoc吸收于400uL的二氯甲烷中,并且用400uL二异丙胺处理。将该混合物搅拌2h,转移到具有甲醇的圆底烧瓶中并且浓缩。将该材料用庚烷(2mL)处理并且浓缩,将该顺序用异丙醇重复以去除过量的二异丙胺。将该材料在高真空下浓缩过夜并且进行到下一步骤。分析MS:m/z(ES+)计算1136.7(M+H)+,观察到1137.6。将粗MMAF-VCP-NH2吸收于DMF(420uL)中,并且用炔烃碳酸酯(40mg/mL,50uL)以及三乙胺(2.8uL)的溶液处理。将该混合物在室温下搅拌8h。分析MS:m/z(ES+)计算1312.8(M+H)+,观察到1313.7。制备紫杉醇-环辛炔衍生物。将紫杉醇(500mg,590umol)以及戊二酐置于50mL的具有磁力搅拌的圆底烧瓶中,并添加吡啶(10mL)。将该溶液搅拌过夜。将该混合物浓缩至油,并且通过硅胶柱色谱法(己烷/丙酮洗脱)纯化,给予该所希望的产物。分析MS:m/z(ES+)计算968.0(M+H)+,观察到969.1。将紫杉酚-戊二酸共轭物于DMF中的溶液(15.6ug/uL,321uL,5mg,5.2umol)置于具有磁力搅拌棒的小瓶中。连续地添加HATU偶联剂的溶液(46.1ug/uL,50uL,2.3mg,6.2umol)、环辛炔-胺的溶液(34ug/uL,50uL,1.7mg,5.2umol)以及三乙胺(1.6uL,11.4umol)。将该小瓶加盖关上并且搅拌过夜。分析MS:m/z(ES+)计算1273.6(M+H)+,观察到1274.4。制备阿霉素-环辛炔衍生物。将阿霉素溶液(12.5ug/uL,320uL,4mg,7.4umol)置于具有小的磁力搅拌棒的小瓶中。将环辛炔碳酸酯于DMSO中的溶液(28.5ug/uL,63uL,1.8mg,7.4umol)添加到该小瓶中。添加三乙胺(2.2uL,16umol),将该小瓶加盖,并且将该反应搅拌4h。分析MS:m/z(ES+)计算719.7(M+H)+,观察到720.5。在重链的位置274处掺入nnAAlys-叠氮化物的抗-Her2抗体(抗-Her2-Lys叠氮化物274h)与MMAF-环辛炔衍生物的共轭在200uLPCR管中放置磷酸盐的溶液(50mM,pH=7.4,3uL)以及抗-Her2抗体(抗-Her2-Lys叠氮化物274h)的溶液(11.43uL,2.1mg/mL)。向其中添加MMAF-炔烃衍生物(1.1uL,14.5mMol)的DMSO溶液,将该管加盖并涡旋。允许该混合物静置4h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,6.4uL)处理,涡旋并且允许静置60min。然后将该混合物通过ZEBA(皮尔斯(Pierce))迷你旋转柱脱盐,以给予最终的ADC溶液(0.21mg/mL)。使用基于细胞的荧光测定以显示该4D5IgG以及共轭的4D5-MMAF结合且内化到表达Her2表位的细胞中。使被用于Her2的表达的构建体稳定转染的乳腺癌细胞系A345或SKBR3以及EL4以及EL4细胞生长于完全RPMI-1640或DMEM中。将细胞进行解离、计数并通过离心收获。对于每次测定,将大约200,000-500,000个细胞用包含0.5%BSA的PBS在室温孵育1小时。然后在37oC,在存在或不存在0.1%叠氮化钠的情况下,将细胞用1ug经纯化的4D5IgG或4D5IgG-MMAF共轭物处理1小时。将细胞进行洗涤并且在37oC与抗-人类IgG-藻红蛋白质共轭物孵育1小时,用PBS洗涤,并且重悬浮于PBS或包含50%台盼蓝的PBS中,并且通过流式细胞术(Accuri)进行分析。细胞生活力和细胞死亡测定。使用MTS测定评估抗-Her2-MMAF共轭物对肿瘤细胞生活力的影响。简而言之,将细胞铺板于96孔板(SKBR3,MDA-MD,以及MCF7的5000个细胞/孔)中于50uL的缺少酚红并包含10%胎牛血清(FBS)的RPMI1640中。在37oC,在5%CO2的湿润环境中,将在50uL的包含FBS的RPMI1640中的不同浓度的抗体共轭物以及对照添加到这些细胞中,持续三天。通过添加20uL的完全MTS(皮尔斯)分析细胞生活力,并且允许在37oC显色1-3小时。使用读板仪(分子动力学公司(MolecularDynamics))记录每个孔在490nm处的吸光度(图8B)。抗-Her2抗体(抗-Her2-Lys叠氮化物274h)与MMAF-缬氨酸-瓜氨酸-p-氨基-苯甲酰基-碳酸酯-环辛炔衍生物的共轭在200uLPCR管中放置磷酸盐的溶液(50mM,pH=7.4,3uL)以及抗-Her2抗体(在重链的位置274处掺入的NNAAlys-叠氮化物)的溶液(11.43uL,2.1mg/mL)。向其中添加MMAF-炔烃衍生物(1.23uL,13mMol)的DMSO溶液,将该管加盖并涡旋。允许该混合物静置4h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,6.4uL)处理,涡旋并且允许静置60min。然后将该混合物通过ZEBA(皮尔斯(Pierce))迷你旋转柱脱盐,以给予最终的ADC溶液(0.21mg/mL)。抗-Her2抗体(抗-Her2-Lys叠氮化物274h)与紫杉醇-环辛炔衍生物的共轭在200uLPCR管中放置磷酸盐的溶液(50mM,pH=7.4,3uL)以及抗-Her2抗体(抗-Her2-Lys叠氮化物274h)的溶液(11.43uL,2.1mg/mL)。向其中添加紫杉醇-炔烃衍生物(1.24uL,12.9mMol)的DMSO溶液,将该管加盖并涡旋。允许该混合物静置4h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,6.4uL)处理,涡旋并且允许静置60min。然后将该混合物通过ZEBA(皮尔斯(Pierce))迷你旋转柱脱盐,以给予最终的ADC溶液。实例9:抗-Her2抗体(抗-Her2-Lys叠氮化物274h)的PEG化20K线性PEG-环辛炔对PEG化至抗-Her2抗体(抗-Her2-Lys叠氮化物274h)在200uLPCR管中放置磷酸盐缓冲液(50mM,pH=7.4,1uL)。添加包含抗体的叠氮化物的溶液(AzAb-2,2.1mg/mL,1.07uL),随后添加20KPEG环辛炔的溶液(60mg/mL,1.0uL)。将该溶液在漩涡器(费希尔(Fisher))上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置4h。将该溶液用水(7uL)稀释,以将最终体积变成~10uL。然后将该溶液分配成5uL并且添加到5uL的还原或非还原胶装载缓冲液中。将该混合物混合且加热至95C持续3分钟。然后将该样本装载到SDS-PAGE凝胶(4%-20%Tris-Gly,英杰公司)上。SDS-PAGE(还原和非还原)指示,该PEG化伴随着几乎所有起始物被转化为双-PEG化种类的高转化率发生。非还原胶(图9A)泳道2:未经处理的抗-Her2抗体(在重链的位置274处掺入的NNAAlys-叠氮化物),泳道3:用20KPEG线性PEG环辛炔处理的抗-Her2抗体(在重链的位置274处掺入的NNAAlys-叠氮化物)。在PEG处理的叠氮化物-抗体至占优势的单一种类方面观察到明显的分子位移,与包含两个PEG链的分子量位移预期一致。PDSI指示高转化率(93%双PEG化,6.9%单-PEG化,无起始材料)。还原胶(图9B)泳道2:抗-Her2抗体(在重链的位置274处掺入的NNAAlys-叠氮化物),泳道3:用20KPEG线性PEG炔烃处理的抗-Her2抗体(在重链的位置274处掺入的NNAAlys-叠氮化物)。观察到该PEG处理的抗体(泳道3)中的重链的明显分子量位移,说明针对包含叠氮化物的重链的特异性以及该PEG共轭的转化度(96.7%,密度测定法)。20KPEG环辛炔PEG化至具有在重链的位置274以及轻链的位置70处掺入的nnAAlys-叠氮化物的抗-Her2抗体(抗-Her2-Lys叠氮化物274h70l)在200uLPCR管中放置该抗-Her2-Lys叠氮化物274h70l抗体的溶液0.5mg/mL,4.5uL)。添加20KPEG环辛炔(60mg/mL,1.0uL)的溶液并且将该溶液在漩涡器(费希尔(Fisher))上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置18h。将该溶液用水(4.5uL)稀释,以将最终体积变成~10uL。然后将该溶液分配成5uL并且添加到5uL的还原或非还原胶装载缓冲液中。将该凝胶样本混合且加热至95C持续3分钟。然后将该样本装载到SDS-PAGE凝胶(4%-20%Tris-Gly,英杰公司)上。SDS-PAGE(非还原)指示,该PEG化伴随着几乎所有起始抗体被转化为四-PEG化种类的高转化率发生。非还原胶(图10A)泳道2:未经处理的抗-Her2抗体(抗-Her2-Lys叠氮化物274h70l),泳道3-5:都用20K线性PEG环辛炔处理,泳道3:抗-Her2抗体(抗-Her2-Lys叠氮化物274h),泳道4:抗-Her2抗体(抗-Her2-Lys叠氮化物274h),泳道5:赫塞汀(无叠氮化物)阴性对照,泳道6:未经处理的抗-Her2抗体(抗-Her2-Lys叠氮化物274h)以及泳道7:未经处理的赫塞汀。从未经处理的4D5AzAb-4的单带至占优势的四-PEG化种类观察到明显分子量位移。这个四-PEG化种类大于双-PEG化种类(泳道4)。该非还原胶也显示了该反应针对包含叠氮化物的抗体的特异性,而不包含叠氮化物的赫塞汀不显示反应性。PDSI指示高转化率(86%双PEG化,14%三-PEG化,无起始材料)。还原胶(图10B)泳道2:未经处理的抗-Her2抗体(抗-Her2-Lys叠氮化物274h70l),泳道3-5:都用20K线性PEG环辛炔处理,泳道3:抗-Her2抗体(抗-Her2-Lys叠氮化物274h70l),泳道4:抗-Her2抗体(抗-Her2-Lys叠氮化物274h),泳道5:赫塞汀(无叠氮化物)阴性对照,泳道6:未经处理的抗-Her2抗体(抗-Her2-Lys叠氮化物274h)以及泳道7:未经处理的赫塞汀。该还原胶显示,重链以及轻链(泳道3)两者都经历了明显分子位移,与将单20KPEG链添加至抗体的每个亚基中一致。这些带是清楚的,指示该反应仅发生在叠氮化物位点,并且没有另外的PEG化发生,如通过不存在另外的更高的MW带的情况指示的。与在该重链的相同位置共享叠氮化物的抗-Her2抗体(抗-Her2-Lys叠氮化物274h)进行的比较指示针对两条带的相同分子量位移以及通过该凝胶的相同运行时间。当用20KPEG炔烃处理时,包含非叠氮化物的赫塞汀不显示反应性。针对抗-Her2抗体(抗-Her2-Lys叠氮化物274h70l)的共轭效率也是高的,该凝胶显示了未经修饰的重链或轻链的极少迹象或无这样的迹象。实例10:通过铜催化点击进行的Her2抗体的PEG化20KPEG炔烃PEG化至抗-Her2抗体(抗-Her2-Lys叠氮化物274h)在200uLPCR管中放置三[(1-苄基-1H-1,2,3-三唑-4-基)甲基]胺的二氯甲烷溶液(TBTA,10mM,1.5uL)。在氮气流下蒸发溶剂,并且添加4D5AzAb-2的溶液(3.5mg/mL,2.14uL)。添加20KPEG炔烃的水性溶液(60mg/mL,1.67uL),随后添加半胱氨酸的水性溶液(20mM,0.5uL)。最后,添加硫酸铜的溶液(10mM,0.75uL),并且将该混合物温和涡旋以混合组分,然后允许静置4h。将该溶液分流(2.5uL),一半添加到7.5uL的还原胶装载缓冲液中,并且一半添加到7.5uL的非还原胶装载缓冲液中。将该样本加热到95C持续3min并且然后装载到SDS-PAGE凝胶(4%-20%Tris-Gly,英杰公司)上并且运行。非还原胶(图11B)泳道2:未经处理的抗-Her2抗体(抗-Her2-Lys叠氮化物274h),泳道3:用20KPEG线性PEG炔烃处理的抗-Her2抗体(抗-Her2-Lys叠氮化物274h)。在该非还原胶(泳道3)中观察到未反应的抗-Her2抗体(在重链的位置274掺入NNAAlys-叠氮化物),被鉴别为包含一条20KPEG链的更高的分子量带以及被鉴别为双-PEG化种类更高的分子量带的混合物。PDSI指示单以及双PEG化种类之间的适度转化。(5.3%双PEG化,37%单-PEG化,58%未经修饰的Ab)。还原胶(图11A)泳道2:未经处理的抗体,泳道3:用20KPEG线性PEG炔烃处理的抗体。通过PDSI进行的凝胶分析指示适量(10%)的重链PEG化。该反应对该重链是特异性的,因为该轻链显示未改变。图32中证明了在CuAAC条件下利用TBTA条件进行的4D5-AzAb(HC274)的20kDaPEG化的另外的实例。当与未经处理的AzAb进行比较时,该PEG化以位点特异性方式发生到具有叠氮化物的重链上,如通过此带的显著分子量位移指示的。20K线性PEG-环辛炔PEG化至在位置117处具有NNAA取代的抗-PSMAscFv(抗-PSMAscFV-117)通过将抗体J591(BANDER)的CDR接枝到scFv框架上产生针对PSMA的scFv。通过基因合成使用重叠寡聚物以及PCR产生针对PSMA的scFv,并且将该产物克隆到pJ201中以产生pJ201-PSMA。通过限制酶消化(XhoI以及NotI)切割该scFv的ORF并且将该DNA片段纯化产生表达构建体pCDNA3.1-PSMA。将质粒pCDNA3.1用相同的酶剪切,并且使用T4DNA连接酶插入该PSMAscFv片段,以产生包含在CMV启动子的控制下的针对PSMA的scFv并且在框架中包含3’5xPro-6xHis标签(编码PPPPPHHHHHH,SEQID81)的pCDNA3.1-J591scFv。为了将琥珀密码子掺入此scFv,使用定点诱变以在该scFv的最后的3’密码子之后但在该5xPro-6xHis标签之前插入琥珀终止密码子。将所得到的构建体命名为抗-PSMAscFV-117。通过DNA测序鉴别包含该琥珀密码子的克隆。包含nnAA的抗-PSMAscFv-117的瞬时表达是在稳定表达pylRS的HEK293细胞中进行的。这个细胞系是通过转染包含pCEP4(生命技术公司)中的pylRS基因的载体并且通过生长于包含潮霉素DMEMB(DMEM(DMEM(生命技术公司),2mMglutamax,1mM丙酮酸钠,6mM谷氨酰胺,1x非必要氨基酸(GibcoCAT#11140-050),10%胎牛血清,以及0.2mg/mL潮霉素)的培养基进行选择产生的。通过限制稀释克隆存活的细胞,并且扩展表明pylRS的高功能活性的克隆。这是通过在存在ALOCnnAA的情况下用编码tRNApyl的载体以及在位置Y40包含琥珀密码子的报告子构建体GFPY40瞬时转染不同克隆来实现的。使用Accuri流式细胞仪在这些细胞中定量荧光水平并且分离高功能性克隆。抗-PSMAscFv117的表达是使用标准转染条件进行的。将细胞铺板至大约90%汇汇合且在37℃生长。翌日,根据具体制造商说明,将这些铺板的细胞与适当的DNA进行孵育,该DNA之前用亲脂性试剂(脂质体转染(Lipofectamine)2000,293fectin(英杰公司)处理过。在存在1-2mMLys-叠氮化物的情况下生长2-5天后,收获该生长培养基,并且直接使用,或将所表达的蛋白质纯化。简而言之,在真核细胞的瞬时表达之后从生长培养基纯化此处所述的表达的scFv。在每种情况下,将0.1体积的10xPBS添加到表达上清液,以平衡这些盐以及该样本的pH。在4oC将表达上清液用PBS透析持续16h。通过批结合或重力流使蛋白质结合到镍-NTA珠(GE医疗公司)上,并且用洗涤缓冲液(50mM磷酸钠pH7.4,300mMNaCl,20mM咪唑)充分洗涤。将结合的材料进行洗脱(50mM磷酸钠pH7.4、300mMNaCl、250-500mM咪唑)。通过SDS-PAGE以及考马斯亮蓝染色鉴别包含靶蛋白质的级分。汇集峰级分,并且在进一步使用之前用PBS进行透析。在200uLPCR管中,添加在位置117处具有NNAA取代的抗-PSMAscFv的溶液(0.3mg/mL,3.6uL),随后添加20KPEG环辛炔的溶液(60mg/mL,1.0uL)。将该溶液在漩涡器(费希尔(Fisher))上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置4h。将该溶液用还原胶缓冲液(6uL)稀释,以将最终体积变成~10uL。然后将该样本装载到SDS-PAGE凝胶(4%-20%Tris-Gly,英杰公司)上。SDS-PAGE(还原性)指示,该PEG化成功耗尽该抗-PSMAscFv带的大多数。还原胶(图12A)泳道2:未经处理的在位置117处掺入NNAAlys-叠氮化物的抗-PSMA。泳道3:用20KPEG线性PEG环辛炔处理的在位置掺入NNAAlys-叠氮化物的抗-PSMAscFv为了确定J591-scFv是否是功能性的并且结合PSMA,使用基于细胞的荧光测定。使前列腺癌PC3(PSMA阴性的)以及LNCaP(PSMA阳性的)以及乳腺癌细胞系A345生长于RPMI-1640中。将细胞进行解离、计数并通过离心收获。对于每次测定,将500,000个细胞用包含0.5%BSA的PBS在室温孵育1小时。然后将细胞用200uL的pCDNA3.1-J591转染上清液处理并且用PBS洗涤。然后在室温下,将细胞用小鼠抗-6xHIS抗体以1ug/mL(Clontech)孵育1小时。将细胞用PBS洗涤,并且在室温下用藻红蛋白质共轭的抗-小鼠抗体(美天旎公司(Miltenyi))孵育30分钟。将细胞用PBS洗涤并且通过流式细胞术(Accuri)进行分析。伴随以下修饰如上进行内化测定:利用经纯化的PSMA用于内化测定。在与抗-PSMAscFv孵育的过程中,将一组细胞也在4oC用叠氮化钠(0.1%)处理1小时以抑制细胞表面标记的内化。在37℃进行其他孵育。此外,在最终的洗涤之后,将表面染色通过添加台盼蓝进行抑制以淬灭藻红蛋白质信号。在位置117处具有NNAA取代的抗PSMAscFv(抗-PSMAscFV-117)与MMAF-缬氨酸-瓜氨酸-p-氨基-苯甲酰基-碳酸酯-环辛炔衍生物的共轭在200uLPCR管中放置磷酸盐的溶液(50mM,pH=7.4,1uL)以及抗-PSMAscFV-117的溶液(40.5uL,2.1mg/mL)。向其中添加MMAF-缬氨酸-瓜氨酸-p-氨基-苯甲酰基-碳酸酯-环辛炔衍生物的DMSO溶液(3.46uL,13mMol),将该管加盖并且涡旋。允许该混合物静置4h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,20uL)处理,涡旋并且允许静置60min。然后将该混合物通过ZEBA(皮尔斯(Pierce))迷你旋转柱脱盐,以给予最终的scFv-药物共轭物溶液。通过SDS-PAGE(还原)对该反应混合物进行的检查指示主要PSMA带的小的分子量位移,与该药物部分至蛋白质的共轭一致。在PAGE凝胶上的轻微位移也在单独的scFv构建体,28D2上观察到(图12)。实例11:诱饵氨基酸的测试在体外测定中,将有效的dnnAA通过其与高亲和力pylRS底物(lys-叠氮化物)竞争的能力,针对连同报告蛋白而观察到的其减小背景琥珀压制水平的能力并且针对其改善之前针对高pylRS/tRNA活性选择的细胞的生活力以及功能的能力进行鉴别。为了鉴别可有效地针对pylRS结合与lys-叠氮化物竞争的类似物,首先将dnnAA针对在体外功能性测定中的功能进行测试,基于表达WTpylRS以及tRNApyl的细胞在报告蛋白GFPY40中的靶位点处引入nnAA的能力。此报告子在其开放阅读框中包含琥珀密码子,其在不存在琥珀压制的情况下产生非荧光的截短蛋白质。在琥珀压制的事件中,产生了可通过荧光检测方法进行检测的全长GFP蛋白质。在此测定中,将稳定表达pylRS的HEK293细胞用tRNA表达构建体以及GFPY40报告子盒进行瞬时转染。然后在存在或不存在不同浓度的dnnAA(范围从0.5mM至2mM)的情况下,将细胞与0.5mMlys叠氮化物进行孵育。然后通过流式细胞术对GFP荧光水平进行定量。对于此测定,确定每个条件下荧光细胞的比例并且绘图。因为预期了这些dnnAA与该nnAA(lys-叠氮化物)针对pylRS/tRNA结合进行竞争,有效的dnnAA应减少或阻止全长GFP的表达(如上所述,dnnAA可通过该tRNA被递送到琥珀位点,但不能传导蛋白质合成)。测试若干具有式VII的dnnAA:dnnAA式VIIB.4(图13),渐增浓度的该dnnAA导致荧光细胞数目相伴减少,表明剂量相关的影响(图13A以及B)。为了确定该dnnAA是否使能够琥珀密码子通读,将经转染的细胞也单独与该dnnAA孵育,并且通过流式细胞术监测GFP表达,具有式VIIB.4的dnnAA不诱导GFP表达。为了控制这些dnnAA的非特异性影响,也在存在dnnAA的情况下对如下细胞进行监测,这些细胞被缺少琥珀密码子的报告蛋白质转染,从而产生独立于琥珀压制的野生型GFP(GFPwt)(图13A)。具有式VIIB.4的dnnAA(图13A)对蛋白质表达不显示影响,表明通过后者dnnAA对琥珀压制的抑制是特异性的。使用相似的体外测定来估计在表1中列出的具有式VIIB的dnnAA的有效性,其中显示了对所测试的氨基酸的描述以及对这些结果的概述。对于这个测定,通过流式细胞术确定每个样本的几何平均荧光强度并且绘图。对于测试的每个dnnAA,测量经野生型GFP转染的对照细胞作为阳性对照。此外,使用经报告子GFPY40转染并且暴露于0.5mMlys叠氮化物或2mMlys叠氮化物的细胞以确定在不存在抑制剂的情况下的最大GFP表达水平。在每种情况下,观察到强健的GFP表达水平。为了测试是否dnnAA中任一个可减少GFP表达的效力,在存在0.5mM、1mM以及2mMlys叠氮化物的情况下对细胞进行孵育。在所有情况下观察到相伴着增加的dnnAA浓度的GFP表达的减少(图14)。具有式VIIB.1、式VIIB.3以及式VIIB.6的dnnAA显示相对于缺少dnnAA的样本的GFP表达的最大减少(图14A,E,I)。它们对GFP表达的抑制随着渐增浓度的dnnAA增加,表明这些是PylRS的特异性高亲和力底物。具有式VIIB.2以及式VIIB.5的dnnAA也显示伴随着渐增浓度的dnnAA的GFP表达的减少(图14C,G)。然而,GFP表达的相对减少更加低,表明这些氨基酸类似物具有对该pylRS的低亲和力。因此,本发明的dnnAA可与lys-叠氮化物nnAA针对结合至pylRSpylRS进行竞争并且干扰nnAA引入的生理学机制。使用经pylRS/tRNA对以及GFPY40报告子构建体转染的但未暴露于lys-叠氮化物或dnnAA的细胞以确定背景琥珀压制的水平,并且用作阴性对照。在每种情况下,观察到低的GFP表达水平。在存在高亲和力底物的情况下针对dnnAA抑制的竞争性测定鉴别了针对结合至pylRS进行竞争的dnnAA,并且因此是pylRS/tRNA的特异性抑制剂。然而,该诱饵nnAA旨在减少在不存在nnAA的情况下背景琥珀压制的水平。为了确定是否背景琥珀压制水平可被减少,我们在包含这些dnnAA中之一的培养基中孵育包含GFPY40报告子构建体以及pylRS/tRNApyl对的细胞。为此,将稳定表达pylRS的HEK293细胞用tRNA表达构建体以及GFPY40报告盒进行瞬时转染。孵育3天之后,将细胞通过流式细胞术针对GFP的表达进行测定并且确定该样本的几何平均荧光强度。我们之前观察到,包含pylRS/tRNA琥珀压制系统的完全互补体的细胞显示该GFP报告子构建体的可检测表达,该表达高于在缺少该琥珀压制系统的细胞中观察到的。此观察表明,存在源自于pylRS/tRNA对的非正交活性,该非正交活性导致比缺少pylRS/tRNA对的细胞更高的琥珀压制水平。为了鉴别能够减少背景琥珀压制水平的dnnAA,将这些经转染的细胞与这些dnnAA中的每个以0.5mM、1mM以及2mM进行孵育,并且通过流式细胞术测量GFP水平。具有式VIIB.1、式VIIB.3、式VIIB.6、式VIIB.2、以及式VIIB.5的dnnAA都显示该背景琥珀压制水平相对于对照样本(不包含dnnAA的细胞(图14A-J;GFPY40+tRNA-nnAA))的减少(图14D、F、J)。背景琥珀压制依赖性GFP表达的减少是剂量依赖性的并且随着dnnAA浓度增加而被改善。有趣地,在此测定中dnnAA之一,式VIIB.2显示背景琥珀压制的非常有效的抑制,相对于对照样本GFP荧光水平减少57.7%但还未被鉴别为lys-叠氮化物的强竞争者。这表明,具有式VIIB.2的dnnAA可以具有针对pylRS的低亲和力,容易被lys-叠氮化物替代。此特点对于平台开发是有吸引力的特征,因为它使得能够阻抑背景琥珀压制,但在添加强pylRS底物例如lys-叠氮化物后该系统可被活化。这些数据表明,该dnnAA可占据pylRS-tRNA对并且阻止天然氨基酸的琥珀压制。在此测定中,式VIIB.1(62.5%降低)、式VIIB.3(49.7%)、式VIIB.6(32%)、以及式VIIB.5(35%)以及式VIIB.4(46.3%)在减少背景琥珀压制水平上也是有效的。通过GFP表达相对于对照样本(未暴露于dnnAA)的减少定量其效力,并且在表1中总结该数据。表1-具有式VIIB的诱饵nnAA实例12.本发明的诱饵nnAA对平台细胞系生活力的影响为了检查本发明的dnnAA是否能够起作用以改善包含pylRS以及tRNApyl的细胞的生活力,我们监测了稳定表达pylRS以及tRNApyl的细胞系的生长以及生活力。对于此测定,将稳定表达pylRS以及tRNApyl的CHO细胞以及在重链中包含琥珀密码子的针对her2/neu的IgG用于此实验,该琥珀密码子显示有效地将nnAA掺入到表达的IgG从而产生包含nnAA的抗体。尽管具有pylRS/tRNApyl对的高表达水平,当生长在缺少nnAA的培养基中时,这个细胞系具有很健壮的细胞生长特征。包含琥珀密码子的高表达的靶的存在很可能对这些细胞具有保护作用,这是通过为它们提供高水平的琥珀密码子,这些琥珀密码子吸收琥珀压制活性并且保护这些细胞免受在必要基因上的背景琥珀压制的影响。然而,在将nnAA(lys-叠氮化物)添加到生长培养基以及活化该琥珀压制机构后,观察到细胞生长速率上的降低。这就是说,该培养物中的细胞密度似乎保持稳定,表明该琥珀压制机构的活化导致抑制细胞生长影响。为了确定是否dnnAA可解救此影响,使培养在无血清培养基中的细胞生长至0.5x106个细胞/mL的细胞密度,并且随后用0.5mMlys叠氮化物单独或与2mMdnnAA组合处理。经七天每天监测细胞生活力和细胞数目。在添加nnAA之后第3天用lys叠氮化物单独处理的细胞达到略低于1x106个细胞/mL的细胞密度,并且保持在这个密度用于该测定的其余部分(七天)(图15A)。生长的缺乏很可能不归因于生活力的损失,因为该培养物在整个实验中保留了高生活力(~70%有生活力的细胞)(图15B)。用2mM具有式VIIB.4的化合物或2mM式VIIB.1与0.5mMlys-叠氮化物组合处理的培养支持该培养物的继续生长,达到超过1.5x106个细胞/mL并且保留90%的细胞生活力。在该测定的持续时间中,用具有式VIIB.2的dnnAA处理的细胞显示细胞密度超过3x106并且生活力远超过90%。相比之下,在该测定的过程中用式VIIB.3处理的细胞显示降低的细胞生活力(<0.5x106个细胞/mL)以及不良生活力(第6天30%-40%)。这些数据表明具有式VIIB.2、式VIIB.4、以及式VIIB.1的dnnAA阻止了由活化该琥珀压制系统诱发的抑制细胞生长作用。在存在具有式VIIB.2的dnnAA的情况下生长的细胞随着时间的推移显示线性细胞生长并且经这七天达到3x106的细胞密度。这些数据指出具有式VIIB.2的dnnAA为针对pylRS/tRNA功能的lys-叠氮化物的最有力的竞争者。以上数据显示,具有式VIIB.2的dnnAA是pylRS/tRNA的有效抑制剂,并且显示在包含高活性琥珀压制机构并且在存在nnAA的情况下表达靶基因的细胞中减少了琥珀压制依赖性细胞生长抑制的影响。然而,dnnAA的预期用途是在具有高活性琥珀压制机构的细胞的发育以及分离过程中保护这些细胞。因此,我们接下来提问,是否dnnAA可改善富含高活性琥珀压制机构的细胞池(平台细胞系)的生活力以及性能。为此,在存在或不存在dnnAA的情况下,使针对该琥珀压制机构的高活性选择的平台细胞系生长若干代,并且随后以十个细胞/孔接种到96-孔板中,并且在存在或不存在dnnAA的情况下生长(式VIIB.2)。将每个板孵育若干天并且将细胞收获、汇集并且计数。有趣地,与诱饵nnAA孵育的板比在不存在dnnAA的情况下生长的那些显示更高的细胞数目(不存在dnnAA时1.66x106以及1.5x106个细胞/mL并且在dnnAA中生长的培养物中3.0以及3.7x106个细胞/mL)。细胞数目两倍的增加可归因于dnnAA的保护作用。为了检查在这些条件下生长的细胞的活性,将从每个板中汇集的0.5x106个细胞接种到6-孔板,并且在不存在dnnAA并且具有lys-叠氮化物的情况下用GFPY40报告子构建体转染。24小时之后,通过流式细胞术分析每个样本中的细胞的荧光强度。将该数据设门控到包括单一细胞并且绘图以展示每个事件的强度(散点图,图16)。针对每个样本确定落到GFP强度谱靠前的10%内的事件的数目(表2)。在存在dnnAA的情况下生长的细胞显示更高数目的落到建立的门控内的事件(无诱饵时n=139并且有诱饵时n=175(式VIIB.2)。这表明,通过生长于包含诱饵nnAA的培养基中保留了更高活性的细胞。利用另外的度量通过分离具有最高的GFP表达的靠前300个事件(前300)的几何平均数来定量细胞的性能。在此度量下,dnnAA孵育的细胞显示超过在不存在dnnAA的情况下生长的细胞的性能改善(无诱饵时GM=954;有诱饵时GM=1142(式VIIB.2)。将来自两个组的细胞也用编码野生型GFP的构建体转染。在这个组上进行相同的分析。将这些数据在表2中进行总结,并且显示生长在包含dnnAA(式VIIB.2)的培养基中的细胞,相对于在不存在dnnAA的情况下生长的相同细胞系,显示更高数目的高荧光性细胞以及更高的荧光水平。表2-dnnAA(式VIIB.2)增加平台细胞群体中的琥珀压制活性:样本前300(GM)#靠前门控中的事件1-示踪物343256-示踪物529591无诱饵10241533无诱饵8851255诱饵11491796诱饵1135171dnnAA似乎保护了这些平台细胞的生活力,但也保护了具有更高水平的琥珀压制功能性的细胞。为了进一步评估dnnAA对包含高活性琥珀压制系统的平台细胞系的细胞生长特征的影响,我们进行了动力学生长测定。为此,如上所述,在存在或不存在dnnAA的情况下,将该平台细胞系在96-孔板中进行孵育。汇集来自每个板的细胞并且一式三份以1000个细胞/孔接种在96孔板中,并且将细胞孵育四天。在第四天,向这些细胞中添加阿尔玛蓝染料,并且通过荧光发射测定生活力。阿尔玛蓝充当细胞生活力的方便的指标。有生活力的细胞代谢阿尔玛蓝产生作为高荧光性染料的试卤灵。在第5、6、9、以及11天监测荧光水平,并且对这些荧光值进行绘图(图17)。与在不存在dnnAA的情况下生长的细胞相比,诱饵生长细胞显示改善的细胞生活力。这是通过在该绘制的生长速率上拟合一条线显示的,并且计算针对每者的斜率。生长于包含诱饵nnAA的培养基中的细胞比在不存在dnnAA的情况下生长的细胞(Avg斜率=669)显示更快的生长速率(Avg斜率=1190)。这些数据显示,使用dnnAA保护了这些细胞使其免受琥珀压制的慢性影响并且改善了该培养物的细胞生活力和生长。这些数据合起来指出,dnnAA是针对开发平台细胞系以及保存具有高琥珀压制活性的细胞的必要组分。实例13:用GFP测定进行的作为nnAA的新颖吡咯赖氨酸类似物的翻译测试。开展基于细胞的体外测定以通过nnAA整合到靶蛋白质的效率评估pylRS/tRNA对和本发明的吡咯赖氨酸类似物(nnAA)的兼容性。为此,使用标准转染方案,将稳定表达pylRS(3H7)的HEK293细胞用质粒瞬时转染用于表达tRNApyl和编码GFPY40(包含琥珀密码子,代替氨基酸残基编号40处的酪氨酸(其中1是起始子蛋氨酸))的报告子构建体。将经转染的细胞与2mM的nnAA孵育2-3天,通过在显微镜下目视检查定性分析GFP产生。通过流式细胞术使用Accuri流式细胞仪量化GFP荧光并确定荧光细胞的几何平均数。使用该基于细胞的测定以确定是否不同的nnAA针对pylRS是适合的底物并允许其翻译为靶蛋白质。在nnAA的存在下孵育表达PylRS/tRNApyl对并包含编码GFPY40报告基因的载体的细胞。容易被PylRS/tRNApyl对利用的nnAA支持将nnAA翻译到GFP的琥珀位点并允许产生全长GFP(荧光蛋白质)的基因的通读。这些细胞的荧光强度依赖于nnAA掺入的效率。因此,可利用性差的nnAA产生弱荧光或不发出发荧光的细胞。显微镜观察鉴别多个可被pylRS使用的nnAA(表3,阳性GFP)。此外,将在每个样品中的相对表达水平与已知可被pylRS有效利用的底物产生的那些相比。式V.1(MFI=931,289),式V.2(MFI=1,676,250)和式V.3(MFI=2,250,000)(参见表3)以几何平均数支持高水平的GFP表达。诸位发明人发现,本发明的类似物式VI.1和VI.3在所使用的实验条件下被掺入到GFP报告基因中并且产生绿细胞。其中,具有式VI.1的类似物支持高水平的GFP表达(MFI904206)并表示在所测试的实验条件下被pylRS/tRNA对高效利用的类似物(参见表4)。表3-式V类似物GFP结果表4-式VI类似物GFP结果抗-Her2抗体的构建和表达在哺乳动物细胞中表达包含两个非天然氨基酸(每个重链中一个)(4D5-2AZab)的全长抗-Her2抗体。将包含叠氮化物部分的nnAA在所选择的位点掺入并且通过亲和色谱法使用蛋白质A树脂(GEHealthcare)或通过IgSelect(通用医疗公司(GEHealthcare),17096901)纯化。然后将经纯化的材料浓缩并经受共轭反应。通过将小鼠抗体4D5的重链和轻链两者的可变区克隆到包含编码人类IgG的基因的载体中产生针对Her2/neu的胞外结构域的抗体。通过基因合成使用重叠寡聚物产生4D5的可变区并将其克隆到由pFUSE-CHIg-hG1(IgG1重链;γ)和pFUSE-CHLIg-hK(轻链;κ;英杰公司)编码的人类IgG1框架中以产生小鼠-人类杂交体。通过定点诱变将琥珀密码子引入到重链(γ)中在位置K274处。通过DNA测序鉴别包含该琥珀密码子的克隆。为了产生整合构建体,将针对该重链的启动子和ORF通过PCR扩增,并通过限制酶消化并连接到pOptivec(生命技术公司(LifeTechnologies))中而克隆。将该轻链和tRNA的单拷贝通过两步骤PCR方法使用重叠寡聚物进行连接,并克隆到可用位点到包含该重链的pOptivec质粒中。然后将该构建体转染到CHO细胞系中,该CHO细胞系包含pylRS/tRNA对和稳定转染的细胞系,该稳定转染的细胞系显示所选择的IgG的高表达。这表示稳定表达包含nnAA的mAb的细胞系的第二个实例,表明该方法对于用在mAb的表达中具有广泛应用性。利用此细胞系来产生包含上述nnAA的IgG。使这些细胞在ExcelDHFR-培养基(西格玛奥德里奇公司(Sigma-Aldrich))中生长到1-2x106个细胞/mL的密度并向培养物中添加nnAA到1mM的最终浓度。将细胞孵育5天并从生长培养基中纯化IgG。收获上清液并使其经受离心以收集悬浮的细胞和其他碎片。然后将上清液通过0.22um过滤器过滤,以在施加到色谱柱之前去除任何颗粒材料。以1-5mL/min流速使用AKTA色谱法系统,将经过滤的上清液施加到1mL-5mL预包装的HiTrap蛋白质A琼脂糖凝胶。将结合的材料和树脂用PBS洗涤以去除松弛结合的蛋白质,并将该结合材料用100mM甘氨酸(pH3.0)以1mL/min的流速洗脱。将包含靶蛋白质的峰级分用0.1体积分数的1MTris-HCl(pH8.0)中和。在4℃,将所有构建体用PBS透析持续16小时并且到最终的磷酸盐缓冲液中。式VI.1作为nnAA在位置274处被掺入到其重链中的抗体被称为“4D5-2AzAb-HC274-(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸”。4D5-2AzAb-HC274-(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸的PEG化在200uLPCR管中放置磷酸盐缓冲液(5uL,500mM,pH=7.4)。添加4D5-2AzAb-HC274-(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸(式VI.1)的溶液(10uL,0.55mg/mL),随后添加20KPEG环辛炔的溶液(3.3,60mg/mL)。将该溶液在漩涡器上大力混合。允许该混合物静置过夜。将该混合物稀释到200uL并施加到蛋白质-A磁珠。将该混合物涡旋并允许旋转以将这些珠混合90min。固定这些珠并处置贯穿的材料。将这些珠用PBS(2X)洗涤并且然后悬浮于还原凝胶缓冲液中。涡旋并且然后加热到95C持续3min。将该悬浮液直接装载到SDS-PAGE凝胶上。SDS-PAGE凝胶的考马斯(Commassie)染色表明该重链的选择性PEG化(图18,泳道3)。通过SPAAC进行的4D5-2AzAb-HC274-(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸与荧光素的共轭。在200uLPCR管中放置磷酸盐缓冲液(65uL,50mM,pH=7.4)。添加4D5-2AzAb-HC274-(2S)-2-氨基-6-{[(2-叠氮基乙基)氨基甲酰基]氧基}己酸的溶液(30uL,0.55mg/mL),随后添加溶液DMCO-Fluor488环辛炔(5.4uL,5mMinDMSO,点击化学工具)的溶液。将该溶液在漩涡器上大力混合。允许该混合物静置24h。将该混合物通过HIC色谱法(TosohTSKgelButylNPR,以1M硫酸钠至磷酸盐缓冲液的梯度)进行分析,显示共轭已经发生并且导致DAR1以及DAR2种类的混合物(图19)实例14:另外的nnAA数据可替代地制备(2S)-2-氨基-6-[[(2-叠氮基乙基)氨基甲酰基]氧基]己酸,式VI.1。步骤1:在具有磁力搅拌器的4mL小瓶中放置Boc-N-6-羟基正亮氨酸(50mg,1eq)和DMF(1mL)。向其中添加2-氯乙基异氰酸酯(17.3mg,1.0eq)和吡啶(32.3uL,2eq)。将该小瓶加盖并允许搅拌5h。将该溶液转移到萃取漏斗,用乙酸乙酯和100mM柠檬酸稀释。摇动该混合物并分离各层。将水层用乙酸乙酯萃取另外两次。将有机层合并,用5%氯化锂洗涤,用硫酸钠干燥,过滤并且浓缩。将该产物直接向前用于下一步骤。分析MS:m/z(ES+)计算352.1(M+H)+,观察到352.1。步骤2:在具有磁力搅拌器的4mL小瓶中放置来自以上的粗的氯衍生物和DMSO(1mL)。将叠氮化钠(130mg,5eq)和吡啶(32.3uL,2eq)添加到该混合物中并将该小瓶加盖。将该混合物在60℃下搅拌过夜。将该混合物转移到萃取漏斗并用100mM柠檬酸和乙酸乙酯稀释。摇动该混合物并分离各层。将水层用乙酸乙酯萃取另外两次。将有机层合并,用5%氯化锂洗涤,用硫酸钠干燥,过滤并且浓缩。将该产物转到下一步骤。分析MS:m/z(ES+)计算359.2(M+H)+,观察到360.2。最终步骤:在20mL小瓶中放置粗的Boc保护的氨基酸和乙腈(2mL)。向其中添加盐酸于二噁烷中的溶液(4N,2.5mL)。将该溶液搅拌2h,并且然后在减压下浓缩。将该混合物冻干为半固体并用在翻译测试中。分析MS:m/z(ES+)计算259.1(M+H)+,观察到260.2。制备(2S)-2-氨基-6-{[(丙-2-烯-1-基)氨基甲酰基]氨基}己酸,式VI.3。在具有磁力搅拌器的4mL小瓶中放置Boc-N-6-羟基正亮氨酸(50mg,1eq)和DMF(1.5mL)。向其中添加烯丙基异氰酸酯(18.0uL,1.0eq)和吡啶(32.3uL,2eq)。将该小瓶加盖并允许搅拌4h。将该溶液转移到萃取漏斗中,用乙酸乙酯和100mM柠檬酸稀释。摇动该混合物并分离各层。将水层用乙酸乙酯萃取另外两次。将有机层合并,用5%氯化锂洗涤,用硫酸钠干燥,过滤并且浓缩。将该产物通过质谱法鉴别并且直接向前用于下一步骤。分析MS:m/z(ES+)计算330.2(M+H)+,观察到331.3。在20mL小瓶中放置粗的于乙腈(2mL)中的羟基亮氨酸-烯丙基氨基甲酸酯衍生物。向其中添加盐酸于二噁烷中的溶液(4N,2.5mL)。将该溶液搅拌2h,并且然后在减压下浓缩。将该混合物冻干为半固体并用在翻译测试中。将该产物通过质谱法确认。用离子交换色谱法(DOWEX-50)进行另外的纯化。分析MS:m/z(ES+)计算230.1(M+H)+,观察到231.2。实例15:IgG第一稳定细胞系构建表达赫塞汀、能够在位置274引入NNAA的细胞系。将DG44CHO细胞用两种载体转染,一种包含针对pOptivec中的重链的表达盒,并且一种针对赫塞汀的pcDNA3.1中的轻链(hygro+)并且包含在位置H274处的琥珀密码子。将细胞在包含潮霉素的培养基中进行选择,并且随后针对表达通过生长于包含甲氨蝶呤的培养基进行选择。通过克隆分离高表达截短IgG的克隆。将最好表达的克隆用编码pylRS以及18个拷贝的U6-tRNApyl(pMOAV-2puro)的载体转染。将经转染的细胞凭借抗生素抗性进行选择,并且通过ELISA测定鉴别显示最高琥珀压制活性的细胞,在将克隆暴露于nnAA(lys-叠氮化物)之后对其全长IgG表达进行定量。分离显示12ug/mL的稳定的IgG表达的克隆,该IgG包含nnAA。此数据说明了构建能够通过pylRS/tRNA对掺入nnAA的表达mAb的细胞系的第三个实例。此外,此途径不同于之前利用的按引入这些功能性元件的顺序的方法。实例16:对于引入nnAA的IgG位置性突变。实例5描述了在该抗-IL6以及抗Her2抗体的重链位置274引入突变并且将该修饰的抗体成功共轭至各种分子。此处,描述了新的IgG位置性突变体以及DAR2和DAR4ADC的产生,从将突变引入到cDNA上到这些ADC的细胞毒性数据。构建4D5抗Her2抗体,其中在重链位置H274和H359以及轻链L70和L81单独放置琥珀终止密码子。该H274、H359以及L81被表达为单独的突变体,并且在HEK293细胞中H274与L70或L81被表达为双突变体。在稳定表达PylRS的HEK细胞中,使这些4D5突变体与Pyl-tRNA共表达。将上清液在蛋白质A上进行纯化,并且将mAbPEG化并通过PAGE进行分析(图20)。该数据指示,PEG化有效地发生在每个位置,至多个位置的共轭同时发生,如通过该反应混合物中存在的DAR4种类例示的。4D5-AzAb(HC274)以及4D5-4AzAb(HC359)经受了明显的分子量位移,这是SDS-PAGE凝胶中的位点特异性PEG化的结果。同样地,4D5-AzAb(LC81)也显示相似的分子量上的增加,如可在PAGE凝胶上观察的。该重链保持未受影响(尽管由于残余的PEG移动穿过凝胶而变形)。包含四种叠氮化物(4D5-AzAb(HC274-LC81))的HC274以及LC81突变体也容易PEG化并且可通过SDS-PAGE凝胶检测。该重链和轻链都显示显著的分子位移,类似于包含两个叠氮化物的抗体的那些(图20)。位置性突变体的PEG化20K线性PEG-环辛炔PEG化至4D5-AzAb(LC81)在200uLPCR管中,添加4D5-2AzAh(LC81)的溶液(8uL,0.106mg/mL),随后添加20KPEG环辛炔的溶液(2.0uL,60mg/mL)。将该溶液在漩涡器上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置24h并且然后通过SDS-PAGE进行分析(图20)。通过明显分子量位移证明了轻链的修饰,与20kDaMWPEG的掺入一致(泳道7)。20K线性PEG-环辛炔PEG化至4D5-AzAb(HC359)在200uLPCR管中,添加4D5-AzAb(HC359)的溶液(8uL,0.145mg/mL),随后添加20KPEG环辛炔的溶液(2.0uL,60mg/mL)。将该溶液在漩涡器上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置24h并且然后通过SDS-PAGE进行分析(图20)。包含叠氮化物的抗体(叠氮化物在位置359)显示明显的分子量位移,对重链是特异性的,作为位点特异性PEG化的结果(泳道5)。20KPEG环辛炔PEG化至4D5-AzAb(HC274:LC70)在200uLPCR管中放置4D5-AzAb(HC274:LC70)的溶液(2uL,0.47mg/mL)。添加20KPEG环辛炔(1.0uL,60mg/mL)的溶液,并且将该溶液在漩涡器上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置24h并且然后通过SDS-PAGE进行分析(图20)。在PAGE凝胶中,重链和轻链都经历明显的分子量增加,作为特异性地通过共轭而具有单PEG附接位点的结果(泳道6)。细胞毒性剂共轭至位置性突变体4D5-AzAb(LC81)与AF-环辛炔衍生物的共轭。在200uLPCR管中放置4D5-AzAb(LC81)的溶液(150uL,0.106mg/mL)以及AF-环辛炔的DMSO溶液(20uL,0.5mMol),将该管加盖并且涡旋,并且允许静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,20uL)处理,涡旋并且允许静置2h。然后将该混合物通过两个迷你ZEBA(皮尔斯(Pierce))旋转柱脱盐,以给予最终的ADC溶液。将该混合物通过SDS-PAGE(图22)以及HIC色谱法(图21)进行分析。所得到的共轭物在HIC色谱图中呈现为单一种类,并且稍比HC274变体更疏水,如通过停留时间确定的。SDS-PAGE(非还原)指示了MW的微小增加,作为共轭该药物的结果(图22)。4D5-AzAb(HC359)与AF-环辛炔衍生物的共轭在200uLPCR管中放置4D5-AzAb(HC359)的溶液(150uL,0.145mg/mL)以及AF-环辛炔的DMSO溶液(20uL,0.75mMol),将该管加盖并且涡旋,并且允许静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,20uL)处理,涡旋并且允许静置2h。然后将该混合物通过两个迷你ZEBA(皮尔斯(Pierce))旋转柱脱盐,以给予最终的ADC溶液。将该混合物通过HIC色谱法进行分析(图21)。所得到的共轭物在该HIC色谱图中呈现为单一种类,并且显著比HC274变体更疏水,如通过停留时间确定的。SDS-PAGE也指示针对HC359变体在分子量上比亲本抗体更高的带的形成。4D5-AzAb(HC274:LC81)与AF-环辛炔衍生物的共轭在200uLPCR管中放置4D5-AzAb(HC274:LC81)的溶液(150uL,0.187mg/mL)以及AF-环辛炔的DMSO溶液(20uL,1mMol),将该管加盖并且涡旋,并且允许静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,20uL)处理,涡旋并且允许静置2h。然后将该混合物通过两个迷你ZEBA(皮尔斯(Pierce))旋转柱脱盐,以给予最终的ADC溶液。将该混合物通过SDS-PAGE(图22)以及HIC色谱法(图21)进行分析。所得到的共轭物在该HIC色谱图中呈现为主要地单一的种类,并且显著比DAR2HC274更疏水。在非还原条件下在SDS-PAGE中也观察到分子量上的增加。4D5-AzAb(HC274:LC70)与AF-环辛炔衍生物的共轭在200uLPCR管中放置4D5-AzAb(HC274:LC74)的溶液(50uL,0.47mg/mL)以及AF-环辛炔的DMSO溶液(5uL,3mMol),将该管加盖并且涡旋,并且允许静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,20uL)处理,涡旋并且允许静置2h。然后将该混合物通过两个迷你ZEBA(皮尔斯(Pierce))旋转柱脱盐,以给予最终的ADC溶液。将该混合物通过SDS-PAGE进行分析(图22)。在非还原条件下在SDS-PAGE中观察到分子量上的增加。体外细胞毒性活性将如上所述产生的ADC针对在SKOV3以及HCC1954以及PC3肿瘤细胞系中的细胞毒活性进行测试,这些肿瘤细胞系是用于测试抗Her2抗体以及ADC细胞系的活性的标准靶细胞。SKOV3以及HCC1954表达高水平的Her2,而PC3以低水平表达Her2:将该细胞毒活性计算为ADC在体外杀死50%的肿瘤细胞的浓度,如在表5中所述。显著地,赫塞汀单独未对所测试的细胞系中的任一个施加细胞毒性影响。表5-显示EC50(以nM计),其表示药物在体外杀死50%肿瘤细胞的浓度:如在图23D、E、F中所示,对于每个位置性突变体,对DAR2以及DAR4ADC进行了比较。在每个Her2阳性肿瘤细胞系中,该DAR4ADC比DAR2更高效,证实该DAR4比该DAR2ADC递送更多的药物。图23显示了细胞毒性测定,表5中的EC50值源自该测定。图23A显示该4D5-AzAb(HC274)-AF以及4D5-AzAb(HC359)-AFDAR2ADC连同该4D5-AzAb(HC274,LC-70)-AFDAR4ADC在该SKOV3肿瘤细胞系中的肿瘤细胞毒活性。这些细胞耐受单独的赫塞汀,但被在不同的位置共轭了毒素的ADC有效地杀死。清晰地,与被动扩散进行比较,该ADC大大降低了杀死这些肿瘤细胞所需的AF的浓度,推测这是通过有效地将所有的AF直接靶向该细胞。图23B显示该4D5-AzAb(HC274)-AF以及4D5-AzAb(HC359)-AFDAR2ADC连同该4D5-AzAb(HC274,LC-70)-AF在HCC1954细胞中的肿瘤细胞毒活性。类似于SKOV3细胞,HCC1954细胞耐受赫塞汀,但被在不同的位置共轭了毒素的ADC有效地杀死。图23C显示4D5-AzAb(HC274)-AF以及4D5-AzAb(HC359)-AFDAR2ADC连同4D5-AzAb(HC274,LC-70)-AFDAR4ADC在PC3肿瘤细胞系中的肿瘤细胞毒活性,PC3肿瘤细胞系表达非常低水平的Her2并且更加耐受ADC的肿瘤杀死作用,如在此图连同表5中看到的。图23D显示该4D5-AzAb(HC274)-AF以及4D5-AzAb(LC-81)-AFDAR2ADC连同该4D5-AzAb(HC274,LC-81)-AFDAR4ADC在HCC1954细胞、过度表达Her2的肿瘤细胞系中的肿瘤细胞毒活性。如在该图连同表5中看到的,该DAR4ADC比DAR2成分更高效。图23E显示该4D5-AzAb(HC274)-AF以及4D5-AzAb(LC-81)-AFDAR2ADC连同该4D5-AzAb(HC274,LC-81)-AF在SKOV3细胞、过度表达Her2的肿瘤细胞系中的肿瘤细胞毒活性。如在该图连同表5中看到的,该DAR4ADC比DAR2成分更高效。图23F显示该4D5-AzAb(HC274)-AF以及4D5-AzAb(LC-81)-AFDAR2ADC连同该4D5-AzAb(HC274,LC-81)-AF在PC3细胞、表达非常低Her2的肿瘤细胞系中的肿瘤细胞毒活性。如在该图连同表5中看到的,这些ADC对此靶存在极少活性或无活性。实例17:在HC274位置共轭的抗体的药物动力学、稳定性和体内抗肿瘤活性抗-Her2抗体(4D5AzAb(HC274))与DBCO-Fluor488的共轭在配备有磁力搅拌器的1000uLHPLC小瓶中放置磷酸盐的溶液(511uL,50mM,pH=7.4)以及该4D5-AzAb的溶液(164uL,6.87mg/mL)。向其中添加DMSO溶液DBCO-Fluor488(75uL,10mM于DMSO中),将该管加盖并且搅拌24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,50uL)处理,并且搅拌2h。然后将该混合物通过ZEBA(皮尔斯)2mL旋转柱脱盐,以给予最终的抗体-染料共轭物。通过HIC色谱法以及SDS-PAGE评估该材料。HIC色谱法指示了单一主要种类的形成,与每抗体添加两个染料分子一致(图24)。将大鼠用4D5AzAb(HC274)-DBCO-Fluor488或赫塞汀以1mg/kg剂量IV注射,并且使用抗-IgGELISA将这两种分子的血清水平监测11天。如在图25A中所示,在恒定重链位置274处对IgG的修饰不会影响药代动力学特征曲线,如通过血清水平测量的并且当与未经修饰的赫塞汀进行比较时。针对IgG的大鼠新生儿Fc受体在与人FcRn相同的残基处的识别人IgGFc结构域。修饰的人类IgG,例如本发明的ADC会保留在体内持续延长的时间段,归因于大鼠FcRn的功能,只要该共轭物与该FcRn的相互作用保持完整。这些数据证明,HC-274修饰的IgG在大鼠中保留长的体内半衰期,指示该FcRn相互作用未被该共轭物阻滞。在4D5上的与大鼠FcRn相互作用的相同残基也负责与人FcRn的相互作用。这些数据显示,在HC-274具有共轭物的ADC将与人类FcRn相互作用并因此在人中保持长的半衰期。通过定量ELISA测定,将针对在图25A显示的PK分析而收集的相同血清针对4D5IgG上FITC共轭物的存在进行测试(图25B),其中该抗体是通过涂覆在塑料ELISA孔上的Her2细胞外结构域蛋白质捕获的。孵育之后,洗涤这些孔并且与共轭至HRP的抗-FITC抗体孵育。孵育之后,洗涤这些孔并且添加HRP底物。此ELISA测量在该ADC上剩余的FITC的量,并且被报导为ng/ml的ADC,所有该FITC是完整的,如在图25B中所示。在具有DAR2的血清中的4D5ADC与未经处理的ADC是相同水平,指示未损失FITC。该数据清楚指示,该染料被完全保留并且在该研究的整个11天中在大鼠中是稳定的。D5-AzAb(HC274)与AF-环炔烃衍生物的共轭。在具有磁力搅拌器的1000uLHPLC管中放置磷酸盐的溶液(24uL,50mM,pH=7.4)以及该4D5-AzAb的溶液(149uL,6.87mg/mL)。向其中添加5的DMSO溶液(27.2uL,2mMol),将该管加盖并且涡旋。允许该混合物静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,50uL)处理,涡旋并且允许静置60min。然后将该混合物通过ZEBA(皮尔斯(Pierce))2mL旋转柱脱盐,以给予最终的ADC溶液。将该混合物通过HIC色谱法以及SDS-PAGE进行分析(图26A以及B)。HIC色谱法指示主要种类的形成,与每抗体被添加两个阿里他汀分子一致。SDS-PAGE(还原)指示针对重链的小的分子量位移,与被添加的阿里他汀分子的添加一致。体外活性将4D5-AzAb(HC274)-AF针对其对Her2阳性细胞系的体外效价进行测试。将过表达Her2的细胞系,SKBR3和SKOV3与PC3(表达非常低水平的Her2的细胞系)进行比较。将4D5-AzAb(HC274)-AF与阿里他汀(AF)单独进行比较。该ADC特异性地杀死该SKBR3以及SKOV3细胞,这些细胞过表达Her2但不杀死表达非常少Her2的PC3细胞。此ADC对3靶细胞系的效价显示在表6中。就显示于图27中的细胞毒性数据而计算这些值。虽然该ADC对SKOV3以及SKBR3显示皮摩尔效价,但它对PC3是无活力的,尽管该细胞系对单独阿里他汀是相当敏感的。这证明了这些ADC对表达高水平的Her2的细胞的特异性。表6-4D5阿里他汀共轭物对Her2阳性肿瘤细胞系的体外效价:EC50nMPC3SKOV3SKBR34D5-AF--0.0190.0074阿里他汀33628771体内抗肿瘤活性SKOV3肿瘤细胞系源自于过表达Her2的人类卵巢癌但抗赫塞汀;在scid小鼠中建立源自于SKOV3细胞的肿瘤。这些肿瘤在2周内达到大约100mm3,并且在那时,将该小鼠随机化,并且将一半的这些小鼠(n=4/组)用6mg/kg的该4D5-2AzAb(HC274)-AFADC的单一皮下注射进行处理(图28)。肿瘤进展之后,将该肿瘤尺寸用卡尺测量。在短期的肿瘤收缩之后,所有经处理的小鼠在肿瘤进展方面显示高显著延迟。一只小鼠被完全治愈,而其他三只小鼠最终复发(图28B)。持续高达80天监测肿瘤进展,以确保该单独的治愈的小鼠不复发。此实例证明,4D5-AFADC被保留在体内循环中,对代谢性降解稳定,但可用以递送高效毒性活性特异性至靶肿瘤细胞的细胞质。实例18:-双特异性抗体/抗体片段的产生以及4D5AzAb-0.5KPEG中间体的表征制备。在200uLPCR管中放置磷酸盐缓冲溶液(34.3uL,50mM,pH=7.4)。向其中添加4D52-AzAb(HC274)的溶液(23.61uL,13mg/mL)以及双-环辛炔连接体的溶液(2.04uL,20mM于二噁烷中,500kDa)。将该混合物经24h时间段间歇涡旋。将该混合物通过CHT树脂纯化,以给予官能化的中间体。28D2scFv-AHA是源自于实例4中的抗-IL6抗体。完全SEQID以及制备的说明可在WO12032181中发现。简而言之,该抗体片段,28D2scFv-AHA表达于大肠杆菌中,并且该nnAA叠氮基高丙氨酸在该序列的C-末端掺入。从该发酵分离该蛋白质,并且通过镍亲和色谱法纯化。制备4D5AzAb(HC274)-28D2(scFv)双特异性物在200uLPCR管中放置4D5-0.5KPEG共轭物(37.5uL,2mg/mL)以及28D2scFv-AHA的溶液(22uL,4.6mg/mL)。将该混合物加盖,涡旋并且允许静置24h。将该混合物通过蛋白质A磁珠(GE)纯化,通过SDS-PAGE进行分析(图29)。SDS-PAGE指示两个独特的带的形成,这两个带的分子量比起始的包含叠氮化物的抗体高,这与一个和两个scFv分子的添加一致。在该ELISA测定中,使抗IgG抗体附于固体表面上。在该双特异性物的Fc区域俘获了Her2-抗-IL6双特异性物。然后将该双特异性物针对功能通过添加IL-6进行评估,转而通过生物素标记检测到该IL-6。在一个ELISA测定中,探测该双特异性物结合该Her2抗原的能力。观察到该双特异性物与对照4D5AzAb(HC274)抗体具有相似的抗原亲和力水平(图30C)。在第二ELISA测定中,探测了该双特异性物结合至IL-6的能力。在该ELISA的这个版本中,该双特异性物是在该ELISA板上通过抗-IgG相互作用捕获的。然后调查IL-6亲和力。发现该双特异性物与全长抗体,13A8具有针对IL-6的相似的亲和力水平(图30B)。在最终的ELISA测定中,探测了该双特异性物在两端在相同时间发挥作用的能力。该双特异性物是通过针对该Her2抗原的抗体亲和力在ELISA板上捕获的。然后探测该IL-6活性。发现该双特异性物同时具有针对Her2抗原以及IL-6的高亲和力。该对照抗体不能显示相似的双功能性活性(图30A)。该FGF21多肽(FGF21—AHA(s86))表达于大肠杆菌中并在SEQID62的位置86修饰以掺入叠氮基高丙氨酸来替代丝氨酸。该修饰的蛋白质被分离为内含体并且通过镍亲和力色谱法纯化。制备4D5AzAb-FGF21(细胞因子)双特异性物在200uLPCR管中的是抗体-连接体中间体的溶液(3.0uL,2mg/mL)以及FGF21-AHA(S86)的溶液(28.6uL,2.8mg/mL)。将该管加盖并且在37C孵育24h。将该混合物通过蛋白质A磁珠纯化并通过SDS-PAGE进行分析(图31)。在该SDS-PAGE凝胶中观察到分子量位移,与通过该中间体连接体的方式将该FGF21(S86)分子添加到该重链一致。实例19.用铜促进的叠氮化物炔烃环加成以及三(3-羟丙基三唑基甲基)胺(THPTA)配体PEG化4D5Azab用THPTA进行20kDAPEG化。在200uLPCR管中放置磷酸盐缓冲液溶液(3uL,150mM,pH=7.4)。向其中添加包含4D5叠氮化物的抗体)6.5uL,4.6mg/mL)的溶液以及20kDaPEG炔烃的溶液(4uL,60mg/mL)。在分开的管中放置硫酸铜的溶液(2.0uL,10mM)以及THPTA(2.5uL,40mM)、氨基胍(1.0uL,100mM)和抗坏血酸钠(1.0uL,100mM)的溶液。将该管加盖,涡旋并且允许静置10min。将全部铜复合体添加到该AzAb-炔烃溶液中。将该最终混合物加盖,涡旋并且允许在37℃或50℃孵育2h。将该反应通过SDS-PAGE进行分析(图33)。SDS-PAGE指示该重链的分子量位移,与添加20kDaPEG一致。用THPTA进行2kDAPEG化。在200uLPCR管中放置包含4D5叠氮化物的抗体(4.9uL,13mg/mL)的溶液以及20kDaPEG炔烃(4.2uL,20mM)的溶液。在分开的管中放置硫酸铜的溶液(3.5uL,20mM)以及THPTA(8.8uL,40mM)、氨基胍(3.5uL,100mM)和抗坏血酸钠(3.5uL,100mM)的溶液。将该管加盖,涡旋并且允许静置10min。将该铜复合体的一部分(1.93uL)添加到该AzAb-炔烃溶液中。将该最终混合物加盖,涡旋并且允许在37℃或60℃孵育2h。将该反应通过SDS-PAGE(图34)以及HIC色谱法进行分析。SDS-PAGE还原显示该重链的分子量上的微小增加,与添加2kDa分子量PEG一致。在非还原条件下SDS-PAGE指示该全长抗体的小的分子量位移,与添加PEG一致。通过指示单一主要种类的HIC色谱法(图34B)提供进一步的证实,与将PEG添加至该抗体一致。实例20.制备另外的细胞毒素-炔烃衍生物。制备鹅膏蕈碱-环辛炔衍生物将ɑ-鹅膏蕈碱(5mg,5.5umol)以及戊二酐(1.5mg,13.2umol)以及吡啶(500uL)置于具有磁力搅拌器的2mL小瓶中。将该溶液搅拌过夜。将该混合物在真空下浓缩,吸收于小量的二氯甲烷中并且沉淀于甲基叔丁醚中。将这些固体转到下一步。分析MS:m/z(ES+)计算1031.4(M+H)+,观察到1033.3。将鹅膏蕈碱-GA(5.6mg,5.5umol)、HBTU(4.6mg)、环辛炔-胺(1.8mg)以及三乙胺(1.9uL)置于5mL离心管中,并溶解于DMF(1mL)中。添加小的磁力搅拌棒并且将该混合物搅拌过夜。从甲基叔丁醚沉淀该混合物。通过离心分离这些固体。分析MS:m/z(ES+)计算1337.6(M+H)+,观察到1339.4。制备阿里他汀F-环辛炔衍生物在具有磁力搅拌器的4mL小瓶中放置于DMF(1mL)中的阿里他汀F(AF)(5mg,10.umol)。向此混合物中添加HBTU(7.6mg,20umol)、BCN胺(3.2mg,10umol)以及三乙胺(3.4uL,25umol)。将该小瓶加盖,并允许将该混合物搅拌过夜。将该溶液通过反相HPLC纯化(乙腈/水0.1%TFA梯度)。汇集所希望的级分并且冻干至粉末。分析MS:m/z(ES+)计算1051.7(M+H)+,观察到1052.7。阿里他汀F-炔丙基酰胺衍生物AF-PA0AF-PA0是指该炔烃以及阿里他汀F之间的PEG间隔子。对于AF-PA0,不存在PEG间隔子,因此是零。AF-PA3在炔烃以及阿里他汀F结构之间掺入三个乙烯单元。在4mL小瓶中放置DMF(1mL)中的阿里他汀F(AF)(6.1mg,8.18umol)。向此混合物中添加HBTU(6.2mg,16umol)、炔丙基胺(0.6uL,9umol)以及三乙胺(2.8uL,20umol)的溶液。将该小瓶加盖,并允许将该混合物孵育过夜。将该溶液通过反相HPLC纯化(乙腈/水0.1%TFA梯度)。汇集所希望的级分并且冻干至粉末。分析MS:m/z(ES+)计算782.5(M+H)+,观察到783.3。阿里他汀F-炔丙基酰胺衍生物AF-PA3在具有磁力搅拌器的1mL小瓶中放置于DMF(200uL)中的阿里他汀F(AF)(4.7mg,6.3umol)。向此混合物中添加HBTU(6.0mg,16umol于50uLDMF中)、丙-2-炔-1-基N-{2-[2-(2-氨基乙氧基)乙氧基]乙基}氨基甲酸酯(3.7mg,14umol于100uLDMF中)以及三乙胺(3.7uL,25umol)的溶液。将该小瓶加盖,并允许将该混合物搅拌过夜。将该溶液通过反相HPLC纯化(乙腈/水0.1%甲酸梯度)。汇集所希望的级分并且冻干至粉末。分析MS:m/z(ES+)计算957.6(M+H)+,观察到958.5。实例21:抗-Her2抗体与毒素-炔烃衍生物的共轭4D5-AzAb(HC274:LC70)与AF-环辛炔衍生物的共轭。在200uLPCR管中放置4D5-AzAb(HC274:LC70)的溶液(24uL,0.5mg/mL)以及AF-环辛炔的DMSO溶液(0.8uL,10mMol),将该管加盖并且涡旋,并且允许静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,20uL)处理,涡旋并且允许静置2h。然后将该混合物通过ZEBA(皮尔斯(Pierce))旋转柱脱盐,以给予最终的ADC溶液。将该混合物通过SDS-PAGE进行分析。将4D5-AzAb(HC274:LC70)-AF以及4D5-AzAb(HC274)-AF针对其杀死Her2阳性细胞的能力通过体外效价测定进行评估。该体外测定描述于实例16中。简而言之,将ADC与未共轭的抗体(赫塞汀)以及游离药物进行比较。在该细胞毒性测定中,发现DAR4ADC比实例15以及图23中所述的相关的DAR2(4D5AzAb(HC274)-AF)化合物对Her2阳性表达细胞系例如SKOV3以及SKBR3稍微更高效。这些化合物对低Her2表达细胞系例如PC3显示最小的活性。两种ADC都比未共轭的抗体或游离药物更高效(图35,A,B,C)。抗-Her2抗体(4D5-AzAb(HC274))与鹅膏蕈碱-环辛炔衍生物的共轭在200uLPCR管中放置磷酸盐的溶液(5uL,50mM,pH=7.4)以及4D5-AzAb(HC274)的溶液(3.94uL,13mg/mL)。向其中添加鹅膏蕈碱炔烃的DMSO溶液(1.13uL,3mMol),将该管加盖并涡旋。允许该混合物静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,7uL)处理,涡旋并且允许静置2h。然后将该混合物通过ZEBA(皮尔斯(Pierce))迷你旋转柱脱盐,以给予最终的ADC溶液4D5-AzAb(HC274)-鹅膏蕈碱。将该4D5-AzAb(HC274)-鹅膏蕈碱针对杀死Her2阳性细胞的能力通过体外效价测定进行评估。该体外测定描述于实例16中。简而言之,将该ADC与另一种ADC(4D5AzAb(HC274)-AF)以及游离药物进行比较。4D5AzAb(HC274)-鹅膏蕈碱ADC在Her2阳性细胞系SKBR3中是有活性的(图36A),而在低Her2表达细胞系例如PC3中显示最小活性(图36B)。4D5-AzAb(HC274)-鹅膏蕈碱与4D5AzAb(HC274)-AF相似地高效,并且比单独游离药物更高效。实例22:用CuAAC进行的4D52AzAb(HC274)至AF-炔烃的共轭4D5-2AzAb(HC274)与AF-PA0或AF-PA3的共轭。在200uLPCR管中放置磷酸盐缓冲液(3.0uL,50-500mM,pH=7.4)、4D5-AzAb(HC274)的溶液(2.1uL,25m/mL在PBS中)以及细胞毒性剂即AF-PA0或AF-PA3的有机溶液(0.70uL,5mM,于DMSO或丙二醇中)。在分开的管中放置硫酸铜(7uL,10-160mM)、THPTA(3.6uL,10-160mM)、氨基胍(7.0uL,10-200mM)、以及抗坏血酸钠(7.0uL,50-300mM)的溶液。将该THPTA-CuSO4复合体加盖,涡旋并且允许静置10min。将该铜复合体(1.23uL/rxn)添加到该AzAb-炔烃溶液中。将该最终混合物加盖,涡旋并且允许(4℃至60℃)孵育0.5-18h。将该材料通过穿过皮尔斯泽巴(PierceZeba)迷你旋转柱(Cat#89882,MWCO=7000)脱盐并且用10XPBS处理。可替代地,将该反应混合物通过CHT色谱法纯化。4D5-2AzAb与AF-PA0的共轭,在室温以1:1THPTA:CuSO4比率。在200uLPCR管中放置磷酸盐缓冲液(2.8uL,500mM,pH=7.4)、4D5-AzAb(HC274)(2.1uL,25mg/mL)以及AF-PA0(0.7uL,5mM,DMSO溶液)。在分开的管中放置硫酸铜(3.5uL,20mM)、THPTA(1.8uL,40mM)、氨基胍(3.5uL,100mM)和抗坏血酸钠(5.3uL,100mM)的溶液。将该THPTA-CuSO4复合体加盖,涡旋并且允许静置10min。将该铜复合体(1.4uL)添加到该AzAb-炔烃溶液中。将该最终混合物加盖,涡旋并且允许在室温孵育1h。将这些反应经由泽巴旋转柱(MWCO=7000)通过脱盐纯化。通过HIC色谱法对该反应的分析指示所希望的DAR2产物的清晰形成。(图38)。通过体外测定将基于CuAAC的抗-Her2阿里他汀ADC与相关的环炔烃衍生的抗-Her2阿里他汀ADC进行比较以测量效价以及选择性。以实例16中所述相似的方式运行体外效价测定。发现基于CuAAC的ADC与该环炔烃衍生的ADC对Her2阳性细胞系例如SKBR3以及SKOV3具有相似的效价(图37A,B)。将相同的ADC针对其对低表达Her2细胞系即PC3的选择性进行测试,并且发现不是高效的(图37C)。表7-针对AzAb共轭利用的CuAAC条件的总结:实例23赫塞汀ADC的产生以及共轭使用实例15中所述细胞系以产生抗-Her2azAb抗体,该抗-Her2azAb抗体源自于赫塞汀(SEQID74以及75,轻链;SEQID76,77,重链),被修饰以在重链(突变体重链,SEQID76,77;未经修饰的轻链根据SEQID78、79)的位置274处包含nnAA,赫塞汀AzAb(HC274)。将3x106个细胞/mL接种到125mL的埃克塞尔(Excell)DHFR-培养基中,并暴露于lys-叠氮化物持续7天。收集该培养基并且通过离心(1000xg持续10min)收获细胞。添加12.5mL的10x磷酸盐缓冲盐水(10xPBS;生命技术公司),并且使培养基三次穿过300ul(包装体积)IgSelect树脂(GE医疗公司)。将该结合的蛋白质用10柱体积的包含0.1%tween-20的Tris缓冲盐水(TBS-TpH7.5)进行洗涤。将赫塞汀-AzAb用0.1M甘氨酸pH2.5洗脱并且收集250ul洗脱级分。将该酸立即用50ul1MTrispH8.0中和。将每个级分通过分光光度法进行分析,并且保留显示OD280读数的级分。合并峰蛋白质级分并且浓缩蛋白质,并且使用Amicon超-4浓缩器(密理博(Millipore))缓冲液交换进磷酸盐缓冲盐水中。处理浓缩的样本用于共轭。20K线性PEG-环炔烃PEG化至赫塞汀-AzAb(HC274)。在200uLPCR管中放置赫塞汀-2AzAb(HC274)(1.0uL,2.4mg/mL)的溶液,随后放置20KPEG环辛炔的溶液(1.0uL,60mg/mL)。将该溶液在漩涡器上大力混合。将该管放置在PCR管上离心数秒,以将所有液体放置到该管的底部。允许该混合物静置4h并且然后通过SDS-PAGE进行分析。SDS-PAGE(还原)指示,该20kDaPEG炔烃以出色的效率位点特异性地共轭至该重链的叠氮化物,具有最少的未反应的重链剩余至无未反应的重链剩余(图39)。赫塞汀-AzAb(HC274)与AF-环辛炔衍生物的共轭。在200uLPCR管放置中该赫塞汀-2AzAb的溶液(19uL,4.8mg/mL)。向其中添加AF-环辛炔衍生物的DMSO溶液(1.5uL,5mMol),将该管加盖并涡旋。允许该混合物静置24h。然后将该反应混合物用叠氮基高丙氨酸的溶液(AHA,250mM于1MHEPES中,10uL)处理,涡旋并且允许静置60min。然后将该混合物通过ZEBA(皮尔斯(Pierce))2mL旋转柱脱盐,以给予最终的ADC溶液。通过HIC色谱法进行的分析指示所希望的DAR2产物的清晰形成(图40)。通过SDS-PAGE(还原)进行的另外的分析指示该重链的分子量的小的增加,非还原PAGE也指示主要的蛋白质带的分子量上的增加。在CuAAC条件下赫塞汀-2AzAb与AF-PA0的共轭。在实例22中所述的条件下进行共轭。将这些反应经由泽巴旋转柱(MWCO=7000)通过脱盐纯化。通过HIC色谱法对该反应的分析指示所希望的DAR2产物的清晰形成(图41)。通过SDS-PAGE(还原)进行的另外的分析指示分子量的小的增加,归因于该药物共轭此亚基。另外的PAGE(非还原)分析也指示该主要蛋白质带的分子量增加(图41)。体外细胞毒性活性将如上所述产生的ADC针对在SKOV3以及PC3肿瘤细胞系中的细胞毒活性进行测试,这些肿瘤细胞系是用于测试抗Her2抗体以及ADC细胞系的活性的标准靶细胞。SKOV3细胞表达高水平的Her2,而PC3细胞以低水平表达Her2。简而言之,对于每次测定,将1000个细胞接种到96孔板的每个孔中,并用滴定的单独阿里他汀F或者通过SPAAC或CUAAC化学产生的赫塞汀-AF共轭物进行孵育。将药物处理的细胞在100ul培养基中在37C孵育3天。将20ul的阿尔玛蓝(生命技术公司)添加到每个孔中,并且将这些细胞孵育16-24小时,并且确定每个孔的OD450nm。这些共轭物的细胞毒活性被计算为在体外杀死50%的肿瘤细胞的ADC浓度,如在表8中所述。表8-赫塞汀ADC对不同的肿瘤细胞系的效价EC50nM如在图42A以及B中所示,将用CUAAC或SPAAC共轭化学构建的赫塞汀-AzAb(HC274)-AFADC进行比较。在每种情况下,该赫塞汀ADC在SKOV3细胞(Her2阳性肿瘤细胞系)中是高效的,但不影响PC3细胞。图42A以及B显示了细胞毒性测定,表8中的EC50值源自该测定。图42A显示该赫塞汀-AzAb(HC274)-CUUAC-AF以及赫塞汀-AzAbSPAAC-AF在该SKOV3肿瘤细胞系中的肿瘤细胞毒活性。这些细胞被通过不同共轭化学产生的共轭了毒素的ADC有效地杀死。清晰地,与被动扩散进行比较,该ADC大大降低了杀死这些肿瘤细胞所需的阿里他汀F的浓度,推测这是通过有效地将所有的AF直接靶向该细胞。图42B显示该赫塞汀共轭物对PC3细胞的影响。此处,SPAAC以及CUUAC两者产生的共轭物在检查的浓度不显示细胞毒性。这些数据显示通过CUUAC以及SPPAAC两种共轭方法产生的赫塞汀ADC的特异性靶向以及活性。参考文献1.布莱特(Blight)等人2004自然(Nature),431333-335(2004)2.陈(Chen),P.,2009阿格纽化学杂志(AgnewChemIntEdEngl.)48,4052-55。3.汉考克(Hancock)等人JACS2010,132,14819-244.赫克特(Hecht)等人,1978JBC253,4517-20。5.赫罗尔德(Herold)等人,2008PNAS105,18507-12。6.卡佛兰(Kavran)等人,2007PNAS104,11268-73。7.科勒(Kohrer)等人,2001PNAS98,14310-15;8.科勒(Kohrer)等人,化学&生物学(Chem&Biol.),(2003)10,1095-1102;9.利伯曼(Liebman)SW.以及谢尔曼(Sherman),F.1976遗传学(Genetics),82,233-249。10.利伯曼(Liebman),SW等人,1976遗传学(Genetics)82,251-272。11.刘(Liu)W.等人2007,自然方法(Naturemethods),4;239-244。12.向井(Mukai)等人2008BBRC371,818-82313.内伊柯瓦(Naykova)等人2003分子进化杂志(JMol.Evol.)57:520-532。14.诺伊曼(Neumann)等人2008自然化学生物学(Nat.Chem.Biol.)4,232-234。15.阮(Nguyen)等人,2009美国化学会志(J.Am.Chem.Soc.)131(25),第8720–8721页16.,野泽(Nozawa)2009,自然(Nature),4571163-67。17.佩蒂特(Pettit)等人,1997有机天然产物化学(Fortschr.Chem.Org.Naturst)70,1-7918.坂本(Sakamoto),K.2002核酸研究(Nucl.AcidRes.)30,4692-4699。19.单(Shan)L.等人,基因医学杂志(JGeneMed.)(2006)8,1400-1406。20.森特(Senter)P.等人,2003血液(Blood)102,1458-65。21.泷本j.2009,分子生物系统(Mol.Biosystems),5,931-34。22.王(Wang)w.自然神经科学(NatureNeuro.)2007,8;1063-1072。23.叶(Ye),S.2008,JBC283,1525-1533。24.柳泽(Yanagisawa)2008化学&生物学(Chem&Biol.)15,1187-1197。25.王(Wang)等人,2011王爱军(AijunWang),纳塔利温布雷德奈恩(NatalieWinbladeNairn),马尔塞洛马雷利(MarcelloMarelli)以及肯尼思格拉博斯坦(KennethGrabstein)(2012),非天然的氨基酸蛋白质工程(ProteinEngineeringwithNon-NaturalAminoAcids),蛋白质工程(ProteinEngineering),普拉温考摩耶(PravinKaumaya)编辑,ISBN:978-953-51-0037-9,英泰(InTech),可获自:http://www.intechopen.com/books/protein-engineering/protein-engineering-with-nonnatural-amino-acids26.费克内尔(Fekner),T.,李(Li),X.,&陈(Chan),M.K.(2010),用于翻译掺入到蛋白质中的吡咯赖氨酸类似物(PyrrolysineAnalogsforTranslationalIncorporationintoProteins),欧洲有机化学杂志(EuropeanJouranalofOrganicChemistry),4171-4179。贯穿本说明书及其跟随的权利要求,除非上下文另有要求,否则词语‘包括’以及变化(如‘已包括’和‘包括了’)应当被理解成是指包含了一个提及的整体、步骤、整体的组或步骤的组,但不排除任何其他整体、步骤、整体的组或步骤的组。在此提及的所有专利以及专利申请均通过引用以其全文结合。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1