一种不饱和透明质酸四糖及其制备方法与流程
本发明属于医药领域,涉及一种四糖化合物,具体涉及一种不饱和透明质酸四糖及其制备方法。
背景技术:
透明质酸(hyaluronicacid,俗称玻尿酸),是一种由d-葡萄糖醛酸和n-乙酰氨基葡萄糖为重复双糖单位,通过β-(1→4)和β-(1→3)糖苷键交替连接而成的大分子酸性黏性多糖,广泛用于医药、化妆品、食品等领域。
目前,质酸四糖通常采用生物提取的方式获得。至1934年meyer等人从牛玻璃球眼中首次提取获得透明质酸,透明质酸的提取源由牛玻璃球扩大至牛睾丸、水蛭等生物体上,通过后续的分离得到纯品;如申请号为201510042090.5的中国发明公开了一种透明质酸四糖和六糖的分离方法,采用q琼脂糖凝胶ff填充的离子交换柱,平衡溶液为ph8~9、50mm以内的tris-hcl缓冲溶液,洗脱溶液为相应ph的1mnacl溶液,通过线性梯度洗脱可将透明质酸四糖和六糖进行分离。
与此同时,以lindahl和choay为首的欧美一些科学家在研究肝素的抗凝、抗血栓作用时,确立了肝素的有效片段是一个含有艾杜糖酸的五糖结构;从那时起,国际上兴起了一个肝素类似结构的糖合成的高潮。如果能够通过化学合成的方法获得上述的透明质酸四糖并改善其抗氧化性,显然具有现实的需求和意义。
技术实现要素:
为克服现有技术中的缺陷,本发明旨在提供一种抗氧化特性的不饱和透明质酸四糖。
为达到上述目的,本发明采用的技术方案是:一种不饱和透明质酸四糖,它是离子化合物且其阴离子结构通式如式(1)所示:
式中,x为氢或甲基。
优化地,它的阳离子为钠离子或氢离子。
本发明的又一目的在于提供一种上述不饱和透明质酸四糖的制备方法,它由式(2)所示结构通式的前驱体经反应获得:
式中,x1和y1相互独立地为烷基酰基或芳香基酰基;x2和x3相互独立地为烷基酰基、芳香基酰基、苄基或取代苄基,x3为烷基酰基、芳香基酰基、苄基或取代苄基,或x2和x3组成烷基缩醛、芳基缩醛或缩酮;x4选自甲基、取代芳基、苄基或取代苄基;y2为烷基或芳香基。
具体地,所述前驱体是将二糖给体和二糖受体进行缩合反应制得:
所述二糖给体的结构通式如式(3)所示,
式中,x5为离去基团;
所述二糖受体的结构通式如式(4)所示,
进一步地,所述离去基团选自三氯亚胺代乙酰基、三氟亚胺代乙酰基、1-戊烯氧基、烷基或芳基硫基。
由于上述技术方案运用,本发明与现有技术相比具有下列优点:本发明不饱和透明质酸四糖,通过将端未葡萄糖醛酸的4,5位设置为不饱和双键,在具有与普通透明质酸同样的强吸水性的同时,还具有更强的抗氧化性。
附图说明
图1为本发明二糖受体的合成路线;
图2为本发明二糖给体的合成路线;
图3为本发明不饱和透明质酸四糖的合成路线;
图4为本发明受体a的甲基糖苷合成路线;
图5为本发明受体a的替代物合成路线。
具体实施方式
本发明不饱和透明质酸四糖,它是离子化合物且其阴离子结构通式如式(1)所示:
式中,x为氢或甲基。
上述不饱和透明质酸四糖的阳离子为钠离子或氢离子。
上述不饱和透明质酸四糖,它由式(2)所示结构通式的前驱体经脱保护基获得:
式中,x1和y1相互独立地为烷基酰基或芳香基酰基;x2和x3相互独立地为烷基酰基、芳香基酰基、苄基或取代苄基,x3为烷基酰基、芳香基酰基、苄基或取代苄基,或x2和x3组成烷基缩醛、芳基缩醛或缩酮;x4选自甲基、取代芳基、苄基或取代苄基;y2为烷基或芳香基。
具体地,所述前驱体是将二糖给体和二糖受体进行缩合反应制得:
所述二糖给体的结构通式如式(3)所示,
式中,x5为离去基团;该离去基团选自三氯亚胺代乙酰基、三氟亚胺代乙酰基、1-戊烯氧基、烷基或芳基硫基;
所述二糖受体的结构通式如式(4)所示,
下面将结合实例对本发明进行进一步说明。
实施例1
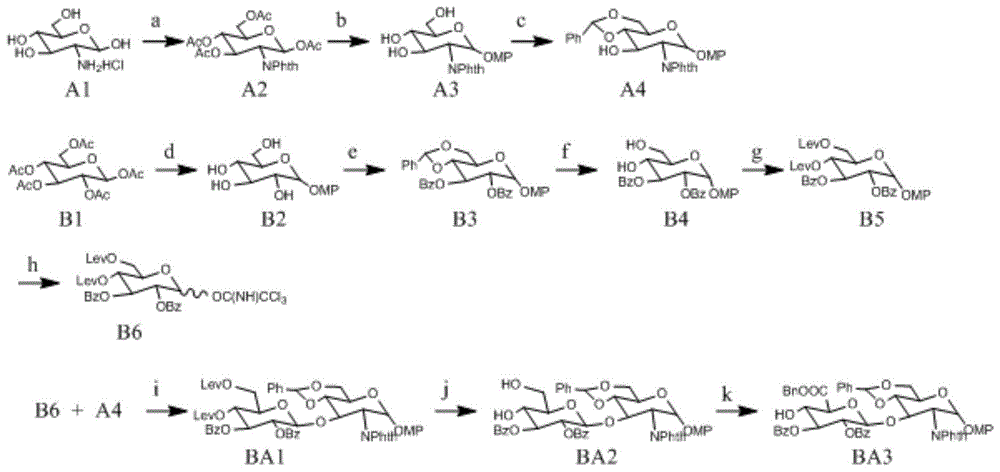
如图1所示,本实施例提供一种二糖受体ba的合成,合成条件a)1.邻苯二甲酸酐,氢氧化钠;2.乙酸酐,dmap,0℃;b)1.对甲氧基苯酚,三氟化硼乙醚,0℃;2.甲醇钠,甲醇;c)苯甲醛二甲缩醛,对甲苯磺酸,dmf;d)1.对甲氧基苯酚,三氟化硼乙醚,0℃;2.甲醇钠,甲醇;e)1.苯甲醛二甲缩醛,对甲苯磺酸,dmf;2.苯甲酰氯,dmap,0℃;f)80%醋酸,70℃;g)乙酰丙酸,碘化2-氯-1-甲基吡啶,dcc,二氯乙烷;h)1.硝酸铈铵,乙腈/水;2.三氯乙腈,dbu,二氯甲烷;i)tmstof,
1、a2的合成
21.5克盐酸氨基葡萄糖溶于150毫升去离子水中,加入60毫升4m氢氧化钠溶液,搅拌30分钟后,分批加入18.2克邻苯二甲酸酐,加完后继续搅拌3小时,过滤,固体用水洗涤后烘干得到粗产品。在此白色固体中加入400毫升二氯甲烷和15克dmap固体,冷却到0℃,滴加50毫升乙酸酐,加完并在此温度下反应1小时后,自然升至室温并继续反应2小时。tlc确认反应完全后,将混合液导入500毫升冰水中,二氯甲烷萃取,将有机相合并后分别用水,饱和碳酸氢钠水溶液,饱和食盐水洗涤,无水硫酸钠干燥,除去有机溶剂后,得到白色固体。白色固体经硅胶柱层析,以乙酸乙酯/正己烷(2:1)洗脱,收集产品部分,旋蒸去除溶剂,得到40.5克a2,收率85%。
2、a3的合成
23.9克a2溶于300毫升干燥二氯甲烷中,加入6.8克对甲氧基苯酚搅拌溶解后,将混合液冷却到0℃,滴加20毫升三氟化硼乙醚溶液。在此温度下反应1小时后自然升温至室温,继续反应3小时。tlc确认反应完成后,将反应混合液倒入冷的饱和碳酸氢钠溶液(300毫升)中,用二氯甲烷萃取。萃取液合并后用水、饱和食盐水洗涤,无水硫酸钠干燥。过滤,旋蒸去除溶剂后得到的产品溶于200毫升甲醇中,加入2克甲醇钠,室温下搅拌3小时至tlc确认反应完全后,加入醋酸中和,旋蒸去除溶剂,得到白色固体。固体用硅胶柱层析,用乙酸乙酯/正己烷(1:1)洗脱,收集产物,去除溶剂后得到16.6克a3,收率80%。
3、a4的合成
15克a3溶于120毫升dmf中,加入15毫升苯甲醛二甲缩醛和1克对甲苯磺酸,在55℃真空下反应3小时后,加入三乙胺中和。混合液经浓缩,硅胶柱层析,以乙酸乙酯/正己烷(1:1)洗脱,去除有机溶剂后,得到15.5克产品a4,收率86%。
1h-nmr(600mhz,cdcl3)δ7.90-7.84(2h,ar-h),7.76-7.72(2h,ar-h),7.52-7.49(2h,ar-h),7.41-7.35(3h,ar-h),6.87-6.83(2h,ar-h),6.76-6.72(2h,ar-h),5.81(1h,h-1),5.60(1h,phch),4.71(1h,h-3),4.51(1h,h-2),4.41(1h,h-6a),3.93-3.83(1h,h-6b),3.76-3.68(5h,h-5,h-4,o-ch3)。
4、b2的合成
20克五乙酰葡萄糖(a1)和7克对甲氧基苯酚溶于150毫升干燥二氯甲烷中,冷却到0℃,滴加20毫升三氟化硼乙醚溶液。滴加完成后,在此温度下反应1小时后自然升温至室温,继续反应3小时。tlc确认反应完成后,将反应混合液倒入冷的饱和碳酸氢钠溶液(300毫升)中,用二氯甲烷萃取。萃取液合并后用水、饱和食盐水洗涤,无水硫酸钠干燥。过滤,旋蒸去除溶剂后得到产品溶于甲醇(200毫升)中,加入1克甲醇钠,室温下搅拌至tlc确认反应完全后,加入醋酸中和,旋转蒸发去除溶剂,得到白色固体。白色固体用硅胶柱层析,用乙酸乙酯/正己烷(1:1)洗脱,收集产物,去除溶剂后,得到14克产物b2,收率95%。
5、b3的合成
将14克b2溶于100毫升干燥的dmf中,加入12毫升苯甲醛二甲缩醛和1克对甲苯磺酸。混合物加热到55℃,真空下反应3小时,tlc确认反应基本完全后,加入三乙胺中和,旋蒸去除溶剂后剩余物中加入100毫升二氯甲烷和8.5克dmap并冷却到0℃,滴加13毫升苯甲酰氯并在此温度下反应30分钟。自然升温至室温,继续反应2小时,tlc确认反应完全后,加入3毫升甲醇,搅拌30分钟。滤除固体后,滤液浓缩得到黄色固体。黄色固体用16毫升乙酸乙酯溶解后加入32毫升正己烷,析出的固体经过滤,烘干,得到21克产物b3。
6、b4的合成
21克b3溶于100毫升氯仿中,然后加入100毫升80%的醋酸溶液。混合液加热到70℃,回流反应至tlc显示反应完全后,旋蒸去除溶剂,残留物用硅胶柱层析,用乙酸乙酯/正己烷(2:1)洗脱,收集产物溶液,旋蒸去除溶剂后得到18克白色固体,收率76%。
比旋光度为+96(20℃,c1.0,chcl3),1h-nmr(600mhz,cdcl3)δ7.98(4h,ar-h),7.55-7.50(2h,ar-h),7.41-7.36(4h,ar-h),6.93-6.89(2h,ar-h),6.80-6.76(2h,ar-h),5.66(1h,h-2),5.47(1h,h-3),5.21(1h,h-1),4.07-4.03(2h,h-6a,h-4),3.94(1h,h6b),3.75(3h,-och3),3.71(1h,h-5)。
7、b5的合成
15克b4溶于20毫升干燥的二氯甲烷和100毫升1,2-二氯乙烷中,加入12.8毫升乙酰丙酸和31.6克碘化2-氯-1-甲基吡啶。搅拌20分钟后,加入21克dcc,继续反应30分钟。tlc确认反应完全后,硅藻土助滤下,去除固体,滤液用500毫升二氯甲烷稀释,用10%的氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋蒸去除有机溶剂。残留物经硅胶柱层析,用乙酸乙酯/正己烷(1:2)洗脱,收集产物,经去除溶剂,得到17.8克产物b5。
8、b6的合成
11.5克b5溶于200毫升乙腈/水(4:1)中,冷却到0℃后,加入27.5克硝酸铈铵。混合液在此温度下反应30分钟,tlc确认反应完全后,加入1升乙酸乙酯,分别用饱和碳酸氢钠,水洗涤,有机相用无水硫酸钠干燥,旋蒸浓缩,用硅胶柱层析,得到淡黄色固体。固体溶于100毫升干燥的二氯甲烷中,加入9毫升三氯乙腈和1毫升dbu,0oc下搅拌3小时后,滤液浓缩,经硅胶柱层析,用乙酸乙酯/正己烷(1:2)淋洗,收集产物,经旋蒸除去有机溶剂后,得到8.7克b6,收率为74%。1h-nmr(600mhz,cdcl3),δ7.96-7.92(4h,ar-h),7.53-7.49(2h,ar-h),7.40-7.34(4h,ar-h),6.76(1h,h-1)6.06(1h,h-3),5.50(1h,h-2),5.45(1h,h-4),4.38-4.34(1h,h-5),4.34(1h,h-6a),4.24(1h,h-6b)2.84-2.71,2.68-2.64(8h,-coch2ch2),2.21(6h,-coch3)。
9、ba1的合成
5克a4与8.8克b6溶于150毫升干燥的二氯甲烷中,干燥的氮气保护下,加入6.5克
10、ba2的合成
5.3克ba1溶于50毫升乙醇/甲苯(2:1)中,室温下加入1.3克醋酸肼,搅拌3小时,tlc确认反应完全后,旋蒸除去溶剂。残留物溶于50毫升二氯甲烷中,分别用1n盐酸、水和饱和食盐水洗涤,无水硫酸钠干燥,旋蒸去除有机溶剂。残留物经硅胶柱层析,以乙酸乙酯/正己烷(2:1)洗脱,收集产物,去除溶剂后得到3.9克产物ba2,收率88%。1h-nmr(600mhz,cdcl3),δ7.96-7.86(6h,ar-h),7.78-7.72(2h,ar-h),7.55-7.47(4h,ar-h),7.42-7.33(7h,ar-h),6.89-6.83(2h,ar-h),6.78-6.73(2h,ar-h;1h,hb-1),6.05(1h,hb-3),5.80(1h,ha-1),5.62(1h,phch),5.50(1h,hb-2),5.45(1h,h-4),4.71(1h,ha-3),4.51(1h,ha-2),4.38-4.34(3h,ha-6a,hb-5,hb-6a),4.24(1h,h-6b),3.93-3.83(1h,ha-6b),3.76-3.68(5h,ha-5,ha-4,o-ch3)。
11、ba3的合成
2.7克ba2溶于40毫升乙腈/水(4:1),室温下加入85毫克tmpo,剧烈搅拌下分批加入2.4克二乙酸碘苯,反应3小时后tlc确认反应完全,真空下旋转蒸出溶剂,加入3次5毫升甲苯并蒸出。残留物溶于25毫升干燥dmf,加入1毫升溴苄和2克碳酸钾,室温下搅拌过夜。过滤除去固体后,真空下蒸出dmf,在剩余物中加入100毫升二氯甲烷,分别用水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,蒸出有机溶剂。残留物经硅胶柱层析,用乙酸乙酯/正己烷(1:2)洗脱,收集洗脱液,旋蒸去除溶剂后得到2.6克白色固体产物ba3,收率88%。1h-nmr(600mhz,cdcl3),δ7.96-7.86(6h,ar-h),7.78-7.72(2h,ar-h),7.55-7.47(6h,ar-h),7.42-7.33(9h,ar-h),6.89-6.83(3h,ar-h),6.78-6.73(2h,ar-h);6.67(1h,hb-1),6.01(1h,hb-3),5.80(1h,ha-1),5.62(1h,phch),5.50(1h,hb-2),5.45(1h,hb-4),5.34(2h,ph-ch2),4.71(1h,ha-3),4.51(1h,ha-2),4.40-4.35(3h,ha-6a,hb-5,hb-6a),4.24(1h,h-6b),3.93-3.83(1h,ha-6b),3.76-3.68(5h,ha-5,ha-4,o-ch3)。
实施例2
本实施例提供一种二糖给体dc的合成,如图2所示,合成条件:a)溴化苄,氢化钠,dmf;b)1.硝酸铈铵,乙腈/水;2.三氯乙腈,dbu,二氯甲烷。
1、合成dc1
2.7克ba2溶于40毫升乙腈/水(4:1),室温下加入85毫克tmpo,剧烈搅拌下分批加入2.4克二乙酸碘苯,反应3小时后tlc确认反应完全,真空下旋转蒸出溶剂,加入3次5毫升甲苯并蒸出。残留物溶于25毫升干燥dmf,加入1毫升溴苄和2克氢化钠(60%nah/重油),室温下搅拌30分钟后,加入200毫升二氯甲烷稀释后加入少量甲醇淬灭后,用水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,有机相经旋蒸除去溶剂后得到淡黄色固体,不经纯化,直接用于下一步反应。
2、合成dc2
上步dc1溶于50毫升乙腈/水(4:1)中,冷却到0℃后,加入5克硝酸铈铵。混合液在此温度下反应30分钟,tlc确认反应完全后,加入200毫升乙酸乙酯,分别用饱和碳酸氢钠,水洗涤,有机相用无水硫酸钠干燥,旋蒸浓缩,用硅胶柱层析,得到淡黄色固体。固体溶于30毫升干燥的二氯甲烷中,加入2毫升三氯乙腈和0.3毫升dbu,0℃下搅拌3小时后,滤液浓缩,经硅胶柱层析,用乙酸乙酯/正己烷(1:2)淋洗,收集产物,经旋蒸除去有机溶剂后,得到2.0克dc2,收率为68%。1h-nmr(600mhz,cdcl3),δ7.96-7.86(5h,ar-h),7.78-7.72(2h,ar-h),7.55-7.47(2h,ar-h),7.42-7.33(5h,ar-h),6.89-6.83(2h,ar-h),6.78-6.73(2h,ar-h;1h,hb-1),6.05(1h,hb-3),5.80(1h,ha-1),5.65(1h,hb-4),5.62(1h,phch),5.50(1h,hb-2),4.71(1h,ha-3),4.51(1h,ha-2),4.38-4.34(2h,ha-6a,hb-6a),4.24(1h,h-6b),3.93-3.83(1h,ha-6b),3.76-3.68(2h,ha-5,ha-4)。
实施例3
本实施例提供一种透明质酸四糖的合成,如图3所示,合成条件为:a)tmstof,
1、全保护透明质酸四糖dcba1的合成
1.3克二糖给体dc2和1克ba3溶于30毫升干燥二氯甲烷中,加入1.5克
2、中间体dcba2的合成
1克dcba1溶于20毫升乙腈/水(4:1)中,冷却到0℃后,加入1.2克硝酸铈铵。混合液在此温度下反应30分钟,tlc确认反应完全后,加入100毫升乙酸乙酯,分别用饱和碳酸氢钠,水洗涤,有机相用无水硫酸钠干燥,旋蒸浓缩,用硅胶柱层析,得到0.8克淡黄色固体,收率86%。
3、中间体dcba3的合成
0.8克dcba2溶于15毫升80%的醋酸溶液中,将此溶液加热到70℃,搅拌反应3小时后,冷却至室温,加入100毫升乙酸乙酯,依次用水、饱和碳酸氢钠、饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋蒸去除有机溶剂后,得到白色固体dcba3,不经纯化,直接用于下一步。
4、不饱和透明质酸四糖的合成
上一步dcba3溶于50毫升正丁醇中,加入8毫升乙二胺,在氮气保护下加热到90℃,并反应20小时,tlc确认反应完全后,旋蒸除去有机溶剂,并用3次5毫升甲苯共蒸除水。剩余物溶于20毫升干燥的二氯甲烷中,加入0.8克dmap后,冷却至-5℃,滴加3毫升乙酸酐,在此温度下反应1小时后自然升温至室温,继续反应2小时。将混合液倒入预冷的饱和碳酸氢钠水溶液中,用90毫升二氯甲烷分3次萃取,有机相合并后依次用水,10%氯化钠溶液洗涤,无水硫酸钠干燥,旋蒸去除有机溶剂后,得到白色固体。白色固体溶于20毫升thf中,在0℃下加入8毫升1m氢氧化锂溶液,在此温度下反应2小时后,自然升温至室温并继续反应20小时。用1m盐酸中和后,旋蒸去除溶剂,残留物经过葡聚糖凝胶g-10柱层析,收集的产物经冷冻干燥,得到0.3克透明质酸四糖产品,收率76%。
1h-nmr(600hz,d2o)δ5.58(d,1h,d-4),5.14(d,1h,a-1),4.68(d,1h,c-1),4.38-4.54(m,2h,b-1,d-1),4.12(m,1h,d-3),3.97(m,1h,a-6a),3.66-3.86(m,10h,a-3,a-5,a-6b,b-2,b-3,b-5,c-3,c-5,c-6a,d-2),3.35-3.60(m,4h,c-6b,b-4,a-4,c-4),3.22-3.34(m,2h,a-2,c-2),1.85,1.96(s,2xac).13c-nmr(150hz,d2o)δ174.6(2xch3co),173.1(b-cooh),163.1(d-cooh),141.2(d5),109.8(d4),102.8,103.0(b-1,d-1),101.1(c-1),96.9(a-1),58.2(a-2,c-2),23.6(2xch3co)。
实施例4
本实施例提供一种不饱和透明质酸四糖的合成,如图4所示:
先合成a的甲基糖苷,合成条件:a)1.邻苯二甲酸酐,氢氧化钠;2.甲醇,乙酰氯;b)苯甲醛二甲缩醛,对甲苯磺酸,dmf。
1.1、合成a2’
21.5克盐酸氨基葡萄糖溶于150毫升去离子水中,加入60毫升4m氢氧化钠溶液,搅拌30分钟后,分批加入18.2克邻苯二甲酸酐,加完后继续搅拌3小时,过滤,固体用水洗涤后烘干得到粗产品。在此白色固体中加入400毫升甲醇,再加入10毫升乙酰氯,加热回流16小时。将反应液旋蒸除去溶剂,并用甲醇和水反复蒸出体系中含有的少量盐酸,得到a2’产物,不经纯化,直接用于下一步反应。
1.2、合成a3’(参照a3->a4的合成)
将上步的a2’溶于250毫升dmf中,加入30毫升苯甲醛二甲缩醛和2.5克对甲苯磺酸,在55℃真空下反应3小时后,加入三乙胺中和。混合液经浓缩,硅胶柱层析,以乙酸乙酯/正己烷(1:1)洗脱,去除有机溶剂后,得到18.5克产品a3’,从盐酸氨基葡萄糖到a3’的收率为45%。
1h-nmr(600mhz,cdcl3)δ7.88-7.81(2h,ar-h),7.73-7.71(2h,ar-h),7.52-7.49(2h,ar-h),7.41-7.35(3h,ar-h),5.81(1h,h-1),5.60(1h,phch),4.71(1h,h-3),4.51(1h,h-2),4.41(1h,h-6a),3.93-3.83(1h,h-6b),3.76-3.68(2h,h-5,h-4),3.38(3h,o-ch3)。
再采用a3’代替图1中a3进行后续的反应(即实施例1-4中的合成工艺过程),得到以下结构的不饱和透明质酸四糖,该不饱和透明质酸四糖与端位为羟基的不饱和透明质酸四糖(即实施例3的产物)在理化性质和生化性质上没有严格意义上的区别,但采用甲基封端可以以达到减少副产物的目的
实施例5
本实施例提供一种不饱和透明质酸四糖(端位为羟基的不饱和透明质酸四糖)的合成,如图5所示,它与实施例1-3中的合成流程基本一致,不同的是:采用a7代替图1中a4进行后续的反应(产物与实施例3的产物相同,主要是提供了不同的合成路线),合成条件:a)氯乙酰氯,dmap,-10℃;b)1.60%醋酸,70℃;2.乙酸酐,dmap,二氯甲烷;c)硫脲,乙醇。
2.1、a5的合成
50.4克a4溶于500毫升二氯甲烷中,加入18.3克dmap,在氮气保护下溶解后将混合液冷却至-10℃,滴加13毫升氯乙烯氯,在此温度下反应1小时后,自然升温至室温,继续反应1小时。将此混合液倒入冰水中,分出有机相,水相用300毫升二氯甲烷分3次萃取,合并后的有机相依次用饱和碳酸氢钠水溶液、水、10%食盐水洗涤,无水硫酸钠干燥,旋蒸除去有机溶剂后剩余物经硅胶柱层析(乙酸乙酯/正己烷=1:3),得到55.1克白色固体产品a5。
2.2、a6的合成
29.0克a5中加入350毫升60%的醋酸溶液,加热到70℃,并在此温度下反应16小时。冷却并浓缩,在剩余物中加入300毫升二氯甲烷,依次用饱和碳酸氢钠水溶液、水、10%食盐水洗涤,无水硫酸钠干燥。在干燥液中加入19克dmap,冷却至-10℃,滴加12.5毫升乙酸酐,并在此温度下反应1小时后,自然升温至室温,继续反应6小时。将反应液倒入冰水中,分出有机相,水相用150毫升二氯甲烷分3次萃取,合并后的有机相依次用饱和碳酸氢钠水溶液、水、10%食盐水洗涤,无水硫酸钠干燥,过滤,旋蒸除去有机溶剂后得到白色固体物a6,不经纯化直接用于下一步反应。
2.3、a7的合成
上步粗产品溶于300毫升乙醇中,加入7.6克硫脲,加热回流2.5小时后,旋蒸除去溶剂。残留物经硅胶柱层析(乙酸乙酯/正己烷=1:2),旋蒸去除有机溶剂后,得到15.5克白色固体a7,步骤2和3两步收率为62%。1h-nmr(600mhz,cdcl3)δ7.52-7.49(2h,ar-h),7.41-7.35(2h,ar-h),6.87-6.83(2h,ar-h),6.76-6.72(2h,ar-h),5.81(1h,h-1),4.68(1h,h-3),4.53(1h,h-2),4.43(1h,h-6a),3.87(1h,h-6b),3.76-3.68(5h,h-5,h-4,o-ch3),2.01(6h,2xac)。
实验例
将实施例3和实施例4中合成的不饱和透明质酸四糖进行抗氧化试验,具体如下:
方法:用胰酶细胞消化液消化hcec细胞,待细胞消化完成后析出消化液,将细胞悬浮于含10%fbs的rmpi1640培养基中并吹成单细胞悬液。计数并稀释成2×105个/ml,混匀后,以每孔3ml加入细胞培养板。接种完成后,将细胞置于37℃、5%二氧化碳培养箱中培养。
将h2o2用rmpi1640配成20mmol/l的溶液,在此溶液中依次加入本申请中的不饱和透明质酸四糖,使得浓度为1、10、100mg/ml,然后分别加入到预先用pbs洗过的培养的细胞,每管1ml,37℃下孵育40分钟。用染色法检测。检测结果,细胞内活性氧(ros)随着不饱和透明质酸四糖的增加,ros的水平下降,在100mg/ml时已经基本接近没有用h2o2处理的正常细胞,即不饱和透明质酸四糖具有明显的抗氧化特性。
对照组:同样的方法将不饱和透明质酸四糖(即本四糖)换成透明质酸四糖(普通透明质酸四糖,作为对比),虽然透明质酸四糖也具有一定的抗氧化特性,但在同样条件下,细胞内ros水平明显高于不饱和透明质酸四糖。
hcec细胞内dcf平均荧光强度:正常细胞110,用h2o2处理后的细胞190,1mg/ml本四糖/h2o2处理后的细胞160,1mg/ml本四糖/h2o2处理后的细胞160,10mg/ml本四糖/h2o2处理后的细胞130,100mg/ml本四糖/h2o2处理后的细胞115;1mg/ml普通透明质酸四糖/h2o2处理后的细胞175,10mg/ml普通透明质酸四糖/h2o2处理后的细胞160,100mg/ml普通透明质酸四糖/h2o2处理后的细胞135。
上述实例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!