聚酰亚胺前体、由其得到的聚酰亚胺树脂膜、以及含有其的显示元件、光学元件、受光元件、触摸面板、电路基板、有机EL显示器、及有机EL元件以及滤色片的制造方法与流程
本发明涉及聚酰亚胺前体、由其得到的聚酰亚胺树脂膜、以及含有其的显示元件、光学元件、受光元件、触摸面板、电路基板、有机EL显示器、及有机EL元件以及滤色片的制造方法。
背景技术:
有机膜与玻璃相比富有弯曲性、不易破裂且轻质。最近,正在研究将有机膜用于平板显示器的基板,将显示器进行挠性化。通常,作为有机膜中使用的树脂,可举出聚酯、聚酰胺、聚酰亚胺、聚碳酸酯、聚醚砜、丙烯酸、环氧树脂等。上述物质中,特别是聚酰亚胺树脂因为在耐热性、机械强度、耐磨耗性、尺寸稳定性、耐药品性、绝缘性等方面优异,所以广泛用于电气·电子产业领域。作为显示器基板,使用聚酰亚胺树脂作为玻璃基板的代替材料时,要求高透明性和低双折射。它们是用于得到鲜明的图像所必需的物性。作为得到透明性高、双折射低的聚酰亚胺的方法,可举出在酸二酐及二胺中,在至少任一方的成分中使用脂环式单体的方法(例如,参见专利文献1)。另一方面,在制造工序中将聚酰亚胺前体树脂组合物涂覆在支承基板上,特别是利用热使聚酰亚胺前体固化而形成聚酰亚胺树脂膜时,存在由于其热应力的原因而使支承基板发生翘曲、聚酰亚胺树脂膜从支承基板剥离之类的问题。为了抑制上述情况,要求聚酰亚胺的热膨胀率接近于支承基板的热膨胀率。但是,大部分的聚酰亚胺的线性热膨胀系数(CTE)在高达50-100ppm/K的范围。近年,也提出了低CTE的聚酰亚胺的方案。有报道指出利用该方案时必要条件是聚酰亚胺的主链结构为直线性且刚直,并且内部旋转被束缚(例如,参见非专利文献1)。具体而言,认为由芳香族酸二酐和芳香族二胺衍生的全芳香族聚酰亚胺是有效的。但是,全芳香族聚酰亚胺在可见光波长区域存在吸收带(其来自于分子内·分子间电荷移动吸收),因此得到的聚酰亚胺树脂膜着色为黄~茶褐色。为了解决该问题,公开了使用了具有脂环式结构(能够赋予透明性)的原料的聚酰亚胺。例如,公开了将刚直结构的脂环式二胺即反式-1,4-二氨基环己烷与刚直的芳香族酸二酐组合而成的聚酰亚胺显示出透明性和低CTE(例如,参见专利文献2)。另外,公开了以含有酯基的脂环式酸二酐作为原料的聚酯酰亚胺显示出高透明性和低双折射(例如,参见专利文献3)。另外,公开了包含3,3’,4,4’-联苯四甲酸二酐、含有芴基的芳香族酸二酐、及反式-1,4-二氨基环己烷的共聚聚酰亚胺显示出高透明性、低双折射、低CTE(例如,参见专利文献4)。现有技术文献专利文献专利文献1:日本特开平11-080350号公报专利文献2:日本特开2012-041530号公报专利文献3:日本特开2007-284414号公报专利文献4:国际公开第2014/007112号非专利文献非专利文献1:Polymer,28,228(1987)
技术实现要素:
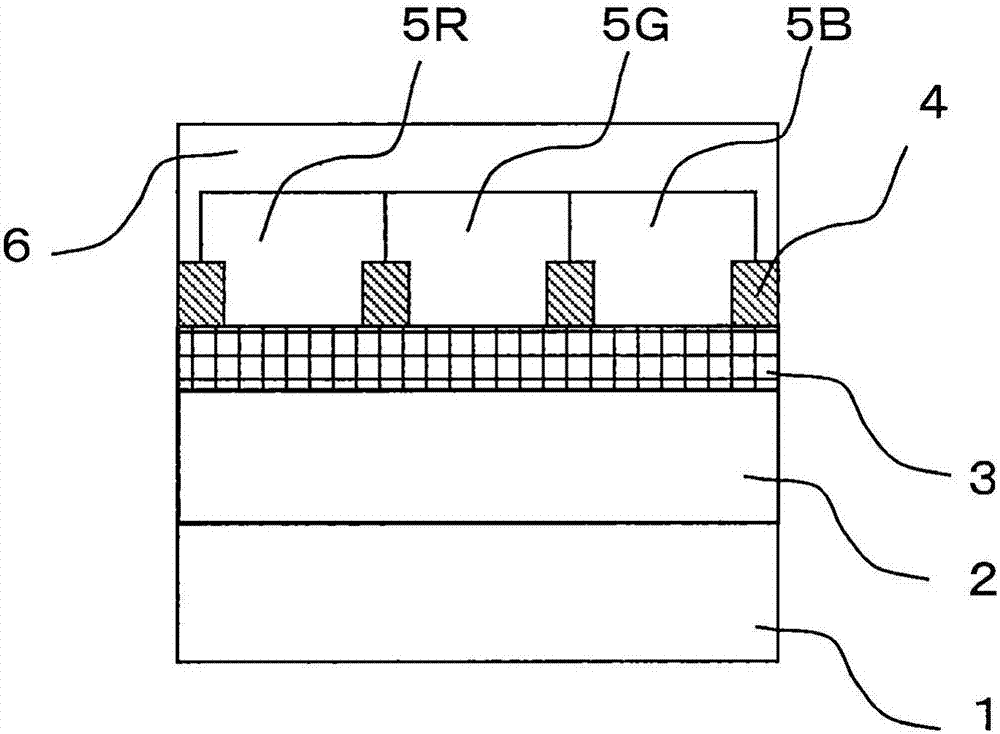
发明所要解决的课题但是,专利文献2中,因为使用刚直结构的单体,所以未赋予低双折射性。进而,专利文献3中记载了除高透明性和低双折射之外,CTE也优选低值,但是对于实现了低双折射的聚酰亚胺而言,CTE在高达70ppm-100ppm/K的范围内,不能同时实现低双折射和低CTE。另外,专利文献4中记载了显示高透明性、低双折射、低CTE,但是就CTE为30ppm/K的低CTE聚酰亚胺而言,双折射为高达0.07以上的值,不能同时实现低双折射和低CTE。如上所述,现状是没有开发出获得了低双折射和低CTE的均衡性的高透明聚酰亚胺。鉴于上述课题,本发明的目的在于提供一种聚酰亚胺前体,所述聚酰亚胺前体经固化后的聚酰亚胺树脂膜具有优异的光透过性、兼备低双折射性和低线性热膨胀性。用于解决课题的手段即,本发明是一种聚酰亚胺前体,其至少含有式(1)表示的酸二酐残基、式(2)表示的二胺残基、以及式(3)表示的二胺残基中的1种以上,相对于聚酰亚胺前体中酸二酐残基的总量,式(1)表示的酸二酐残基为50摩尔%以上,相对于聚酰亚胺前体中二胺残基的总量,式(2)表示的二胺残基为50摩尔%以上,相对于聚酰亚胺前体中二胺残基的总量,式(3)表示的二胺残基为15摩尔%以下。式(3)中的R1~R8分别独立地选自氢、烷基、环烷基、杂环基、链烯基、环烯基、烷氧基、芳醚基、芳基、卤代烷基、氰基及甲硅烷基,它们可以进一步具有取代基,另外,彼此相邻的基团可以具有键而形成稠环结构。式(3)中的A1及A2可以相同也可以不同,为由芳香族环、脂肪族环、链状烃基或者它们的组合形成的结构,或者由它们与选自酰胺基、酯基、醚基、亚烷基、氧化烯基、亚乙烯基及卤代亚烷基中的1种以上的基团的组合形成的结构。发明效果根据本发明能够得到一种聚酰亚胺前体,该聚酰亚胺前体经固化后的聚酰亚胺树脂膜在可见光波长区域显示高透明性、低双折射和低CTE。附图说明[图1A]表示滤色片之一例的剖面图[图1B]表示滤色片之一例的剖面图[图1C]表示滤色片之一例的剖面图[图1D]表示滤色片之一例的剖面图[图2]表示有机EL元件之一例的剖面图[图3]表示有机EL显示器之一例的剖面图[图4]表示滤色片之一例的剖面图[图5]表示滤色片之一例的剖面图[图6]表示滤色片之一例的剖面图[图7]表示有机EL显示器之一例的剖面图[图8]表示有机EL显示器之一例的剖面图[图9]表示有机EL显示器之一例的剖面图具体实施方式以下,详细说明用于实施本发明的方案。需要说明的是,本发明不限于以下实施方式。<聚酰亚胺前体>本发明的聚酰亚胺前体含有式(1)表示的酸二酐残基、式(2)表示的二胺残基、以及式(3)表示的二胺残基中的1种以上,相对于聚酰亚胺前体中酸二酐残基的总量,式(1)表示的酸二酐残基为50摩尔%以上,相对于聚酰亚胺前体中二胺残基的总量,式(2)表示的二胺残基为50摩尔%以上,相对于聚酰亚胺前体中二胺残基的总量,式(3)表示的二胺残基为15摩尔%以下。式(3)中的R1~R8分别独立地选自氢、烷基、环烷基、杂环基、链烯基、环烯基、烷氧基、芳醚基、芳基、卤代烷基、氰基及甲硅烷基,它们可以进一步具有取代基,另外,彼此相邻的基团可以具有键而形成稠环结构。式(3)中的A1及A2可以相同也可以不同,为由芳香族环、脂肪族环、链状烃基、或者它们的组合形成的结构,或者由它们与选自酰胺基、酯基、醚基、亚烷基、氧化烯基、亚乙烯基及卤代亚烷基中的1种以上的基团的组合形成的结构。所述基团中,所谓烷基,是指例如甲基、乙基、正丙基、异丙基、正丁基、仲丁基及叔丁基等饱和脂肪族烃基,其可以具有取代基也可以不具有取代基。被取代时追加的取代基没有特别限定,例如,可举出烷基、芳基及杂芳基等,关于这方面在以下的记载中也共通。所谓环烷基,是指例如环丙基、环丁基、环戊基、环己基、降冰片基及金刚烷基等饱和脂环式烃基,其可以具有取代基也可以不具有取代基。所谓杂环基,是指例如吡喃环、哌啶环、环状酰胺等环内具有碳以外的原子的脂肪族环,其可以具有取代基也可以不具有取代基。杂环基的碳原子数没有特别限定,通常在2以上20以下的范围。所谓链烯基,是指例如乙烯基、烯丙基、丁二烯基等含有双键的不饱和脂肪族烃基,其可以具有取代基也可以不具有取代基。所谓环烯基,是指例如环戊烯基、环戊二烯基、环己烯基等含有双键的不饱和脂环式烃基,其可以具有取代基也可以不具有取代基。所谓烷氧基,是指例如甲氧基、乙氧基及丙氧基等介由醚键键合有脂肪族烃基的官能团,该脂肪族烃基可以具有取代基也可以不具有取代基。所谓芳醚基,是指例如苯氧基等、介由醚键键合有芳香族烃基的官能团,芳香族烃基可以具有取代基也可以不具有取代基。所谓芳基,是指例如苯基、萘基、联苯基、芴基、菲基、三联苯基、蒽基及芘基等芳香族烃基、或者它们多个连接而成的基团,其可以无取代也可以被取代。所述芳基可以具有的取代基为烷基、环烷基、链烯基、炔基、烷氧基、芳醚基、烷基硫基、卤素、氰基、氨基(氨基可以进一步被芳基、杂芳基取代)、甲硅烷基及硼基等。所谓卤代烷基,是指例如三氟甲基之类的、烷基的氢全部或者一部分被卤素取代的基团。所谓甲硅烷基,是指三甲基甲硅烷基、三乙基甲硅烷基之类的、硅的反应点被烷基链取代的基团,或硅的反应点被烷氧基取代的基团。所谓彼此相邻的基团具有键而形成稠环结构,若利用上述式(3)进行说明,则表示选自R1~R8中的任意相邻的2个取代基(例如R3和R4)互相键合形成共轭或非共轭的稠环。上述稠环可以在环内结构中含有氮、氧、硫原子,也可以进一步与其他环键合。另外,式(3)中,所谓A1及A2中的芳香族环,表示由苯、萘、联苯、菲、蒽及芘等芳香族烃衍生的环。所谓脂肪族环,表示由环丙烷、环己烷、降冰片烯及金刚烷等饱和脂环式烃衍生的环。所谓链状烃基,表示直链、或者具有支链的烃基,其中不含有环状烃基。所谓亚烷基,是指例如亚甲基、亚乙基、正亚丙基、异亚丙基、正亚丁基、仲亚丁基及叔亚丁基等二价饱和脂肪族烃基,其可以具有取代基也可以不具有取代基。所谓氧化烯基,是指例如氧化乙烯基、氧化丙烯基等介由醚键键合有脂肪族烃基的二价的官能团,该脂肪族烃基可以具有取代基也可以不具有取代基。所谓亚乙烯基,表示从乙烯中除去2个氢而得的二价基团。所谓卤代亚烷基,表示从卤代烷基中除去1个氢而得的二价基团。相对于聚酰亚胺前体中的酸二酐残基的总量,式(1)表示的酸二酐残基为50摩尔%以上,优选为70摩尔%以上,较优选为80摩尔%以上。由于具有一定量以上的刚直的联苯结构,所以由本发明的聚酰亚胺前体得到的聚酰亚胺显示低CTE性和耐药品性。需要说明的是,式(1)表示的酸二酐残基相对于聚酰亚胺前体中的酸二酐残基的总量的比例的上限为100摩尔%,但是由于还优选含有后述式(4)或式(25)~(28)的酸二酐残基,所以这种情况下,式(1)表示的酸二酐残基相对于聚酰亚胺前体中的酸二酐残基的总量的比例优选为90摩尔%以下。相对于聚酰亚胺前体中的二胺残基的总量,式(2)表示的二胺残基为50摩尔%以上,优选为70摩尔%以上,更优选为80摩尔%以上,特别优选为90摩尔%以上。需要说明的是,因为必须含有式(3)表示的二胺残基,所以式(2)表示的二胺残基相对于聚酰亚胺前体中的二胺残基的总量的比例的上限优选为98摩尔%、较优选为95摩尔%以下。式(2)表示的二胺残基具有脂环式结构,所以通过在分子链中含有一定量以上的式(2)表示的二胺残基,能够缩短分子链内的共轭,将聚酰亚胺前体进行酰亚胺化后的聚酰亚胺显示高透明性。另外,因为是刚直的结构,所以聚酰亚胺显示低CTE。另外,本发明的聚酰亚胺前体中含有式(3)表示的二胺残基。该残基的结构中含有芴结构。通过使芴于9,9位与聚酰亚胺前体的主链键合,由此相对于聚合物主链为垂直方向上的极化率降低,结果酰亚胺化后的聚酰亚胺的双折射降低。另外,式(3)表示的芴残基为刚直的结构,因此来自式(1)表示的联苯基、式(2)表示的环己基的主链的刚直性不会显著降低。其结果,由本发明的聚酰亚胺前体得到的聚酰亚胺显示低CTE。式(3)表示的二胺残基的含量相对于二胺残基的总量而言为15摩尔%以下。从降低相对于聚合物主链为垂直方向的极化率的观点考虑,式(3)表示的二胺残基的含量的下限优选为1摩尔%以上,较优选为3摩尔%以上,更优选为5摩尔%以上。通过式(3)表示的二胺残基在上述范围,低双折射和低CTE的均衡性进一步提高。进而,与式(2)表示的环己基不同,式(3)表示的二胺残基为芳香族二胺残基,所以形成在可见光波长区域具有吸收带的电荷转移络合物。但是,若式(3)表示的二胺残基为上述范围,则可见光波长区域中的透过率不会显著降低。其结果,由本发明的聚酰亚胺前体得到的聚酰亚胺显示高透明性。作为本发明的其他方案,为进一步含有式(4)表示的酸二酐残基中的1种以上的聚酰亚胺前体。式(4)中的R9~R16分别独立地选自氢、烷基、环烷基、杂环基、链烯基、环烯基、烷氧基、芳醚基、芳基、卤代烷基、氰基及甲硅烷基,它们可以进一步具有取代基,另外,彼此相邻的基团可以具有键而形成稠环结构。式(4)中,R9~R16中的烷基、环烷基、杂环基、链烯基、环烯基、烷氧基、芳醚基、苯氧基、芳基、卤代烷基、甲硅烷基、及“彼此相邻的基团具有键而形成稠环结构”的定义与上述式(3)的R1~R8中的上述基团的定义相同(后述的R17~R157也相同)。式(4)中的B1及B2可以相同也可以不同,为由芳香族环、脂肪族环、链状烃基、或者它们的组合形成的结构,或者由它们与选自酰胺基、酯基、醚基、亚烷基、氧化烯基、亚乙烯基及卤代亚烷基中的1种以上的基团的组合形成的结构。式(4)中,B1及B2中的芳香族环、脂肪族环、链状烃基、亚烷基、氧化烯基、亚乙烯基、卤代亚烷基的定义与上述式(3)的A1及A2中的上述基团的定义相同。式(4)表示的酸二酐残基在结构中具有芴结构。如上所述,通过具有芴结构,不会显著降低主链的刚直性,将聚酰亚胺前体进行酰亚胺化后的聚酰亚胺的双折射降低。因此,通过在酸二酐残基和二胺残基两者中导入芴结构,所形成的聚酰亚胺膜的双折射进一步降低。并且,因为是芴部分相对于聚合物主链扭曲的结构,所以分子链内的电荷转移络合物的形成得以抑制。其结果,由本发明的聚酰亚胺前体得到的聚酰亚胺显示高透明性。式(4)表示的酸二酐残基的含量优选相对于聚酰亚胺前体中的酸二酐残基的总量而言为40摩尔%以下。通过为上述范围,低双折射和低CTE的均衡性提高。另外,机械强度优异、减少成膜时的裂缝,故优选。进而,从减少成膜时的裂缝的观点考虑,式(4)表示的酸二酐残基的含量相对于酸二酐残基的总量而言较优选为30摩尔%以下,更优选为25摩尔%以下。另外,含有式(4)表示的酸二酐残基时,作为其含量的下限值,相对于酸二酐残基的总量而言优选为1摩尔%以上,较优选为5摩尔%以上,更优选为10摩尔%以上。本发明的聚酰亚胺前体同时含有式(3)表示的二胺残基和式(4)表示的酸二酐残基时,只要式(1)、(2)、(3)、(4)表示的残基的含量的范围满足上述各自的含有率的范围,则式(3)和式(4)表示的残基的总量没有特别限定。其中,从减少成膜时的裂缝的观点考虑,式(3)和式(4)的残基总量相对于全部残基的含量而言,优选为40摩尔%以下,较优选为30摩尔%以下,更优选为15摩尔%以下。较优选式(3)中的A1及A2分别为由以下(5)~(10)中任一个表示的结构。式(5)~(7)中的X为单键、醚基、亚烷基、氧化烯基或亚乙烯基,一方的键与芴环连接。此处,所谓单键,是指X的两侧不存在原子或连接基团而是直接以单键键合,亚烷基等的定义与上述式(3)的A1及A2的X中的上述基团的定义相同(以后关于Y也相同)。式(8)~(10)中的Y为酰胺基、酯基、醚基、亚烷基、氧化烯基、亚乙烯基或卤代亚烷基。式(5)~(10)中的R17~R80分别独立地选自氢、烷基、环烷基、杂环基、链烯基、环烯基、烷氧基、芳醚基、芳基、卤代烷基、氰基、羟基及甲硅烷基。其中,式(5)中,R17~R21中的任一个为含有X的连接基团。式(6)中,R22~R32中的任一个为含有X的连接基团。式(8)~(10)中,2个环结构中的任一个环可以连接于芴环。另外,R33~R37中的任一个、及R38~R48中的任一个分别为含有Y的连接基团。R49~R53中的任一个、及R54~R58中的任一个分别为含有Y的连接基团。R59~R69中的任一个、及R70~R80中的任一个分别为含有Y的连接基团。较优选式(4)中的B1及B2分别为由以下的(11)~(17)中的任一个表示的结构。式(11)~(13)中的X为单键、醚基、亚烷基、氧化烯基或亚乙烯基,一方的键与芴环连接。式(14)~(17)中的Y为酰胺基、酯基、醚基、亚烷基、氧化烯基、亚乙烯基或卤代亚烷基。式(14)~(17)中的Z为单键,一方的键与芴环连接。式(11)~(17)中的R81~R157分别独立地选自氢、烷基、环烷基、杂环基、链烯基、环烯基、烷氧基、芳醚基、芳基、卤代烷基、氰基、羟基及甲硅烷基。其中,式(11)中,R81~R84中的任一个为含有X的连接基团。式(12)中,R85~R94中的任一个为含有X的连接基团。式(14)中,R98~R102中的任一个、及R103~R112中的任一个分别为含有Y的连接基团。式(15)中,R113~R123中的任一个、及R124~R127中的任一个分别为含有Y的连接基团。式(16)中,R128~R132中的任一个、及R138~R136中的任一个分别为含有Y的连接基团。式(17)中,R137~R147中的任一个、及R148~R157中的任一个分别为含有Y的连接基团。作为式(3)表示的二胺残基的优选例,可举出选自式(18)~(21)中的至少一种的二价有机基团,但不限于此。作为式(4)表示的酸二酐残基的优选例,可举出选自式(22)~(24)中的至少一种的4价有机基团,但不限于此。就本发明的聚酰亚胺前体而言,在不损害本发明的效果的范围,除了至此说明过的残基以外,还可以含有其他成分。所谓其他成分,可举出除了衍生式(1)及式(4)表示的酸二酐残基的酸二酐以外的酸二酐的残基(将这样的酸二酐及其残基分别称作“其他酸二酐”及“其他酸二酐残基”)、除了衍生式(2)及式(3)表示的二胺残基的二胺以外的二胺的残基(将这样的二胺及其残基分别称作“其他二胺”及“其他二胺残基”)等。作为其他酸二酐,可举出芳香族酸二酐、脂环式酸二酐、或脂肪族酸二酐。作为芳香族酸二酐,可举出均苯四甲酸二酐、2,3,3’,4’-联苯四甲酸二酐、2,2’,3,3’-联苯四甲酸二酐、3,3’,4,4’-三联苯四甲酸二酐、3,3’,4,4’-氧邻苯二甲酸二酐(3,3’,4,4’-oxyphthalicdianhydride)、2,3,3’,4’-氧邻苯二甲酸二酐、2,3,2’,3’-氧邻苯二甲酸二酐、二苯砜-3,3’,4,4’-四甲酸二酐、二苯甲酮-3,3’,4,4’-四甲酸二酐、2,2-双(3,4-二羧基苯基)丙烷二酐(2,2-bis(3,4-dicarboxyphenyl)propanedianhydride)、2,2-双(2,3-二羧基苯基)丙烷二酐、2,2-双(4-(3,4-二羧基苯氧基苯基)丙烷二酐、1,1-双(3,4-二羧基苯基)乙烷二酐、1,1-双(2,3-二羧基苯基)乙烷二酐、双(3,4-二羧基苯基)甲烷二酐、双(2,3-二羧基苯基)甲烷二酐、双(3,4-二羧基苯基)醚二酐、1,4-亚苯基双(1,3-二氧代-1,3-二氢异苯并呋喃-5-甲酸酯)、2,2-双(4-(4-氨基苯氧基苯基)丙烷、1,2,5,6-萘四甲酸二酐、2,3,6,7-萘四甲酸二酐、2,3,5,6-吡啶四甲酸二酐、3,4,9,10-苝四甲酸二酐、2,2-双(3,4-二羧基苯基)六氟丙烷二酐、2,2-双(4-(3,4-二羧基苯甲酰氧基)苯基)六氟丙烷二酐、1,6-二氟均苯四甲酸二酐、1-三氟甲基均苯四甲酸二酐、1,6-二(三氟甲基)均苯四甲酸二酐、2,2’-双(三氟甲基)-4,4’-双(3,4-二羧基苯氧基联苯二酐等,但不限于此。作为脂环式酸二酐,可示例1,2,3,4-环丁烷四甲酸二酐、1,2,3,4-环戊烷四甲酸二酐、1,2,4,5-环己烷四甲酸二酐、1,2,3,4-环戊烷四甲酸二酐、1,2,3,4-四甲基-1,2,3,4-环丁烷四甲酸二酐、1,2-二甲基-1,2,3,4-环丁烷四甲酸二酐、1,3-二甲基-1,2,3,4-环丁烷四甲酸二酐、1,2,3,4-环庚烷四甲酸二酐、2,3,4,5-四氢呋喃四甲酸二酐、3,4-二羧基-1-环己基琥珀酸二酐、2,3,5-三羧基环戊基乙酸二酐、3,4-二羧基-1,2,3,4-四氢-1-萘琥珀酸二酐、双环[3,3,0]辛烷-2,4,6,8-四甲酸二酐、双环[4,3,0]壬烷-2,4,7,9-四甲酸二酐、双环[4,4,0]癸烷-2,4,7,9-四甲酸二酐、双环[4,4,0]癸烷-2,4,8,10-四甲酸二酐、三环[6,3,0,0<2,6>]十一烷-3,5,9,11-四甲酸二酐、双环[2,2,2]辛烷-2,3,5,6-四甲酸二酐、双环[2,2,2]辛-7-烯-2,3,5,6-四甲酸二酐、双环[2,2,1]庚烷四甲酸二酐、双环[2,2,1]庚烷-5-羧甲基-2,3,6-三甲酸二酐、7-氧杂双环[2,2,1]庚烷-2,4,6,8-四甲酸二酐、八氢萘-1,2,6,7-四甲酸二酐、十四氢蒽-1,2,8,9-四甲酸二酐、3,3’、4,4’-二环己烷四甲酸二酐、3,3’、4,4’-氧双环己烷四甲酸二酐、5-(2,5-二氧代四氢-3-呋喃基)-3-甲基-3-环己烯-1,2-二甲酸酐、及“Rikacid”(注册商标)BT-100(以上,商品名,新日本理化(株)制)及它们的衍生物等,但不限于此。作为脂肪族酸二酐,可举出1,2,3,4-丁烷四甲酸二酐、1,2,3,4-戊烷四甲酸二酐及它们的衍生物等,但不限于此。所述芳香族酸二酐、脂环式酸二酐、或脂肪族酸二酐可以单独使用或组合两种以上进行使用。上述物质中,从市售并容易获得的观点、从高透明性、低双折射的观点考虑,优选还含有选自2,2-双(3,4-二羧基苯基)六氟丙烷二酐(式(25))、3,3’,4,4’-氧邻苯二甲酸二酐(式(26))、2,2-双(4-(3,4-二羧基苯氧基苯基)丙烷二酐(式(27))、1,2,4,5-环己烷四甲酸二酐(式(28))中的至少一种的酸二酐残基。通过在含有式(1)、(2)、(3)的聚酰亚胺前体中共聚所述酸二酐,即使不使用式(4)表示的含有芴环的酸二酐,聚酰亚胺的双折射也降低。并且,若比较包含式(4)表示的酸二酐残基的酸二酐和式(25)~(28)表示的酸二酐,则后者容易获得、预焙膜的耐裂缝性优异。上述酸二酐的量没有特别限定,从CTE及双折射的观点考虑,优选相对于聚酰亚胺前体中的酸二酐残基的总量而言为10~20摩尔%,从透明性的观点考虑,优选式(3)表示的二胺残基的摩尔数等于或小于选自式(25)~(28)中的至少一种的酸二酐残基的总摩尔数。另外,作为其他二胺,可举出芳香族二胺化合物、脂环式二胺化合物、或脂肪族二胺化合物。例如,作为芳香族二胺化合物,可举出3,4’-二氨基二苯基醚、4,4’-二氨基二苯基醚、3,4’-二氨基二苯基甲烷、4,4’-二氨基二苯基甲烷、3,3’-二氨基二苯砜、3,4’-二氨基二苯砜、4,4’-二氨基二苯砜、3,4’-二氨基二苯基硫醚、4,4’-二氨基二苯基硫醚、1,4-双(4-氨基苯氧基)苯、联苯胺、2,2’-双(三氟甲基)联苯胺、3,3’-双(三氟甲基)联苯胺、2,2’-二甲基联苯胺、3,3’-二甲基联苯胺、2,2’,3,3’-四甲基联苯胺、间苯二胺、对苯二胺、1,5-萘二胺、2,6-萘二胺、双(4-氨基苯氧基苯基)砜、双(3-氨基苯氧基苯基)砜、双(4-氨基苯氧基)联苯、双{4-(4-氨基苯氧基)苯基}醚、1,4-双(4-氨基苯氧基)苯、或者在所述芳香族环上利用烷基、烷氧基、卤素原子等进行取代而得的二胺化合物,但不限于此。作为脂环式二胺化合物,可举出环丁烷二胺、异佛尔酮二胺、双环[2,2,1]庚烷二甲基胺、三环[3,3,1,13,7]癸烷-1,3-二胺、1,2-环己基二胺、1,3-环己基二胺、顺式-1,4-环己基二胺、4,4’-二氨基二环己基甲烷、3,3’-二甲基-4,4’-二氨基二环己基甲烷、3,3’-二乙基-4,4’-二氨基二环己基甲烷、3,3’,5,5’-四甲基-4,4’-二氨基二环己基甲烷、3,3’,5,5’-四乙基-4,4’-二氨基二环己基甲烷、3,5-二乙基-3’,5’-二甲基-4,4’-二氨基二环己基甲烷、4,4’-二氨基二环己基醚、3,3’-二甲基-4,4’-二氨基二环己基醚、3,3’-二乙基-4,4’-二氨基二环己基醚、3,3’,5,5’-四甲基-4,4’-二氨基二环己基醚、3,3’,5,5’-四乙基-4,4’-二氨基二环己基醚、3,5-二乙基-3’,5’-二甲基-4,4’-二氨基二环己基醚、2,2-双(4-氨基环己基)丙烷、2,2-双(3-甲基-4-氨基环己基)丙烷、2,2-双(3-乙基-4-氨基环己基)丙烷、2,2-双(3,5-二甲基-4-氨基环己基)丙烷、2,2-双(3,5-二乙基-4-氨基环己基)丙烷、2,2-(3,5-二乙基-3’,5’-二甲基-4,4’-二氨基二环己基)丙烷、或者在所述脂环上利用烷基、烷氧基、卤素原子等进行取代而成的二胺化合物,但不限于此。作为脂肪族二胺化合物,可举出乙二胺、1,3-二氨基丙烷、1,4-二氨基丁烷、1,5-二氨基戊烷、1,6-二氨基己烷、1,7-二氨基庚烷、1,8-二氨基辛烷、1,9-二氨基壬烷、1,10-二氨基癸烷等烷撑二胺类、双(氨基甲基)醚、双(2-氨基乙基)醚、双(3-氨基丙基)醚等乙二醇二胺类(ethyleneglycoldiamines)、及1,3-双(3-氨基丙基)四甲基二硅氧烷、1,3-双(4-氨基丁基)四甲基二硅氧烷、α,ω-双(3-氨基丙基)聚二甲基硅氧烷等硅氧烷二胺类,但不限于此。所述芳香族二胺化合物、脂环式二胺化合物、或脂肪族二胺化合物可以单独使用或组合两种以上进行使用。上述物质中,从市售且容易获得的观点、及从光学特性的观点考虑,优选2,2’-双(三氟甲基)联苯胺、3,3’-二氨基二苯砜、4,4-二氨基二苯砜、4,4’-二氨基二环己基甲烷。对于本发明的聚酰亚胺前体而言,为了将分子量调整到优选范围,可以利用封端剂将两末端封端。作为与酸二酐反应的封端剂,可举出一元胺、一元醇等。另外,作为与二胺化合物反应的封端剂,可举出酸酐、一元羧酸、单酰氯化合物(monoacidchloridecompounds)、单活性酯化合物、二碳酸酯类、乙烯醚类等。另外,通过使用封端剂,可以导入各种有机基团作为末端基团。作为用于与酸酐基反应的封端剂的一元胺,可举出5-氨基-8-羟基喹啉、4-氨基-8-羟基喹啉、1-羟基-8-氨基萘、1-羟基-7-氨基萘、1-羟基-6-氨基萘、1-羟基-5-氨基萘、1-羟基-4-氨基萘、1-羟基-3-氨基萘、1-羟基-2-氨基萘、1-氨基-7-羟基萘、2-羟基-7-氨基萘、2-羟基-6-氨基萘、2-羟基-5-氨基萘、2-羟基-4-氨基萘、2-羟基-3-氨基萘、1-氨基-2-羟基萘、1-羧基-8-氨基萘、1-羧基-7-氨基萘、1-羧基-6-氨基萘、1-羧基-5-氨基萘、1-羧基-4-氨基萘、1-羧基-3-氨基萘、1-羧基-2-氨基萘、1-氨基-7-羧基萘、2-羧基-7-氨基萘、2-羧基-6-氨基萘、2-羧基-5-氨基萘、2-羧基-4-氨基萘、2-羧基-3-氨基萘、1-氨基-2-羧基萘、2-氨基烟酸、4-氨基烟酸、5-氨基烟酸、6-氨基烟酸、4-氨基水杨酸、5-氨基水杨酸、6-氨基水杨酸、氰尿酰胺(日文原文为“アメライド”)、2-氨基苯甲酸、3-氨基苯甲酸、4-氨基苯甲酸、2-氨基苯磺酸、3-氨基苯磺酸、4-氨基苯磺酸、3-氨基-4,6-二羟基嘧啶、2-氨基苯酚、3-氨基苯酚、4-氨基苯酚、5-氨基-8-巯基喹啉、4-氨基-8-巯基喹啉、1-巯基-8-氨基萘、1-巯基-7-氨基萘、1-巯基-6-氨基萘、1-巯基-5-氨基萘、1-巯基-4-氨基萘、1-巯基-3-氨基萘、1-巯基-2-氨基萘、1-氨基-7-巯基萘、2-巯基-7-氨基萘、2-巯基-6-氨基萘、2-巯基-5-氨基萘、2-巯基-4-氨基萘、2-巯基-3-氨基萘、1-氨基-2-巯基萘、3-氨基-4,6-二巯基嘧啶、2-氨基苯硫酚、3-氨基苯硫酚、4-氨基苯硫酚、2-乙炔基苯胺、3-乙炔基苯胺、4-乙炔基苯胺、2,4-二乙炔基苯胺、2,5-二乙炔基苯胺、2,6-二乙炔基苯胺、3,4-二乙炔基苯胺、3,5-二乙炔基苯胺、1-乙炔基-2-氨基萘、1-乙炔基-3-氨基萘、1-乙炔基-4-氨基萘、1-乙炔基-5-氨基萘、1-乙炔基-6-氨基萘、1-乙炔基-7-氨基萘、1-乙炔基-8-氨基萘、2-乙炔基-1-氨基萘、2-乙炔基-3-氨基萘、2-乙炔基-4-氨基萘、2-乙炔基-5-氨基萘、2-乙炔基-6-氨基萘、2-乙炔基-7-氨基萘、2-乙炔基-8-氨基萘、3,5-二乙炔基-1-氨基萘、3,5-二乙炔基-2-氨基萘、3,6-二乙炔基-1-氨基萘、3,6-二乙炔基-2-氨基萘、3,7-二乙炔基-1-氨基萘、3,7-二乙炔基-2-氨基萘、4,8-二乙炔基-1-氨基萘、4,8-二乙炔基-2-氨基萘等,但不限于此。作为用于与酸酐基反应的封端剂的一元醇,可举出甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、1-戊醇、2-戊醇、3-戊醇、1-己醇、2-己醇、3-己醇、1-庚醇、2-庚醇、3-庚醇、1-辛醇、2-辛醇、3-辛醇、1-壬醇、2-壬醇、1-癸醇、2-癸醇、1-十一烷醇、2-十一烷醇、1-十二烷醇、2-十二烷醇、1-十三烷醇、2-十三烷醇、1-十四烷醇、2-十四烷醇、1-十五烷醇、2-十五烷醇、1-十六烷醇、2-十六烷醇、1-十七烷醇、2-十七烷醇、1-十八烷醇、2-十八烷醇、1-十九烷醇、2-十九烷醇、1-二十烷醇、2-甲基-1-丙醇、2-甲基-2-丙醇、2-甲基-1-丁醇、3-甲基-1-丁醇、2-甲基-2-丁醇、3-甲基-2-丁醇、2-丙基-1-戊醇、2-乙基-1-己醇、4-甲基-3-庚醇、6-甲基-2-庚醇、2,4,4-三甲基-1-己醇、2,6-二甲基-4-庚醇、异壬基醇、3,7-二甲基-3-辛醇、2,4-二甲基-1-庚醇、2-庚基十一烷醇、乙二醇单乙醚、乙二醇单甲醚、乙二醇单丁基醚、丙二醇1-甲基醚、二乙二醇单乙醚、二乙二醇单甲醚、二乙二醇单丁基醚、环戊醇、环己醇、环戊烷单甲醇(cyclopentanemonomethylol)、二环戊烷单甲醇、三环癸烷单甲醇、降冰片(norborneol)、松油醇等,但不限于此。作为用于与氨基反应的封端剂的酸酐、一元羧酸、单酰氯化合物及单活性酯化合物,可举出邻苯二甲酸酐、马来酸酐、纳迪克酸酐、环己烷二甲酸酐、3-羟基邻苯二甲酸酐等酸酐、2-羧基苯酚、3-羧基苯酚、4-羧基苯酚、2-羧基苯硫酚、3-羧基苯硫酚、4-羧基苯硫酚、1-羟基-8-羧基萘、1-羟基-7-羧基萘、1-羟基-6-羧基萘、1-羟基-5-羧基萘、1-羟基-4-羧基萘、1-羟基-3-羧基萘、1-羟基-2-羧基萘、1-巯基-8-羧基萘、1-巯基-7-羧基萘、1-巯基-6-羧基萘、1-巯基-5-羧基萘、1-巯基-4-羧基萘、1-巯基-3-羧基萘、1-巯基-2-羧基萘、2-羧基苯磺酸、3-羧基苯磺酸、4-羧基苯磺酸、2-乙炔基苯甲酸、3-乙炔基苯甲酸、4-乙炔基苯甲酸、2,4-二乙炔基苯甲酸、2,5-二乙炔基苯甲酸、2,6-二乙炔基苯甲酸、3,4-二乙炔基苯甲酸、3,5-二乙炔基苯甲酸、2-乙炔基-1-萘甲酸、3-乙炔基-1-萘甲酸、4-乙炔基-1-萘甲酸、5-乙炔基-1-萘甲酸、6-乙炔基-1-萘甲酸、7-乙炔基-1-萘甲酸、8-乙炔基-1-萘甲酸、2-乙炔基-2-萘甲酸、3-乙炔基-2-萘甲酸、4-乙炔基-2-萘甲酸、5-乙炔基-2-萘甲酸、6-乙炔基-2-萘甲酸、7-乙炔基-2-萘甲酸、8-乙炔基-2-萘甲酸等一元羧酸类及上述羧基进行酰氯化而成的单酰氯化合物、及对苯二甲酸、邻苯二甲酸、马来酸、环己烷二甲酸、3-羟基邻苯二甲酸、5-降冰片烯-2,3-二甲酸、1,2-二羧基萘、1,3-二羧基萘、1,4-二羧基萘、1,5-二羧基萘、1,6-二羧基萘、1,7-二羧基萘、1,8-二羧基萘、2,3-二羧基萘、2,6-二羧基萘、2,7-二羧基萘等二甲酸类的仅单羧基进行了酰氯化而成的一元酰氯化合物、单酰氯化合物与N-羟基苯并三唑、N-羟基-5-降冰片烯-2,3-二羧基酰亚胺进行反应而得到的活性酯化合物。作为用于与氨基反应的封端剂的二碳酸酯化合物,可举出二碳酸二叔丁基酯、二碳酸二苄基酯、二碳酸二甲基酯、二碳酸二乙基酯。作为用于与氨基反应的封端剂的乙烯基醚化合物,可举出氯甲酸叔丁基酯、氯甲酸正丁基酯、氯甲酸异丁基酯、氯甲酸苄基酯、氯甲酸烯丙基酯、氯甲酸乙基酯、氯甲酸异丙基酯等氯甲酸酯类、异氰酸丁酯、异氰酸1-萘基酯、异氰酸十八烷基酯、异氰酸苯基酯等异氰酸酯化合物类、丁基乙烯基醚、环己基乙烯基醚、乙基乙烯基醚、2-乙基己基乙烯基醚、异丁基乙烯基醚、异丙基乙烯基醚、正丙基乙烯基醚、叔丁基乙烯基醚、苄基乙烯基醚等。作为用于与氨基反应的封端剂的其他化合物,可举出氯甲酸苄基酯、苯甲酰氯、氯甲酸芴基甲基酯、氯甲酸2,2,2-三氯乙基酯、氯甲酸烯丙基酯、甲磺酰氯、对甲苯磺酰氯、苯基异氰酸酯等。对于与酸酐基反应的封端剂的导入比例而言,相对于酸二酐成分,优选0.1~60摩尔%的范围,特别优选5~50摩尔%。另外,对于与氨基反应的封端剂的导入比例而言,相对于二胺成分,优选0.1~100摩尔%的范围,特别优选5~90摩尔%。可以通过使用多种封端剂而导入多种末端基团。导入到聚酰亚胺前体中的封端剂可以通过以下方法容易地检测。例如,将导入了封端剂的聚合物溶解在酸性溶液中,分解为作为聚合物的结构单元的胺成分和酸酐成分,通过对其进行气相色谱(GC)测定、NMR测定,能够容易地检测封端剂。除此之外,通过对导入有封端剂的聚合物进行热解气相色谱(PGC)测定、红外吸收光谱测定、13CNMR光谱测定,可以容易地检测。本发明的聚酰亚胺前体优选制成含有溶剂的聚酰亚胺前体树脂组合物。作为溶剂,可以单独使用或使用两种以上的下述物质:N-甲基-2-吡咯烷酮、γ-丁内酯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N,N-二甲基亚丙基脲、1,3-二甲基-2-咪唑啉酮、二甲基亚砜等极性非质子性溶剂、四氢呋喃、二氧杂环己烷、丙二醇单甲基醚等醚类、丙酮、甲基乙基酮、二异丁基酮、二丙酮醇等酮类、乙酸乙酯、丙二醇单甲基醚乙酸酯、乳酸乙酯等酯类、甲苯、二甲苯等芳香族烃类等。相对于聚酰亚胺前体100重量份,聚酰亚胺前体树脂组合物中的溶剂的含量优选为50重量份以上,较优选为100重量份以上,优选为2,000重量份以下,较优选为1,500重量份以下。若为50~2,000重量份的范围,则成为适合涂布的粘度,能够容易地调节涂布后的膜厚。聚酰亚胺前体树脂组合物可以含有表面活性剂。作为表面活性剂,可举出Fluorad(商品名,住友3M(株)制)、Megafac(商品名,DIC(株)制)、Surflon(商品名,旭硝子(株)制)等氟系表面活性剂。另外,可举出KP341(商品名,信越化学工业(株)制)、DBE(商品名,ChissoCorporation制)、Glanol(商品名,工荣社化学(株)制)、BYK(BYKJapanKK制)等有机硅氧烷表面活性剂。Emulmin(三洋化成工业(株)制)等聚氧亚烷基十二烷基醚、聚氧乙烯十二烷基醚、聚氧乙烯油基醚及聚氧乙烯十六烷基醚表面活性剂。进而,可举出Polyflow(商品名,工荣社化学(株)制)等丙烯酸聚合物表面活性剂。相对于聚酰亚胺前体树脂组合物中的聚酰亚胺前体100重量份,表面活性剂优选为0.01~10重量份。聚酰亚胺前体树脂组合物可以含有内部脱模剂。作为内部脱模剂,可举出长链脂肪酸等。就聚酰亚胺前体树脂组合物而言,为了提高与支承基板的粘合性,在不损害保存稳定性的范围,可以含有三甲氧基氨基丙基硅烷、三甲氧基环氧基硅烷、三甲氧基乙烯基硅烷、三甲氧基硫醇丙基硅烷等硅烷偶联剂。优选含量为,相对于聚酰亚胺前体100重量份而言为0.01~5重量份。聚酰亚胺前体树脂组合物可以含有热交联剂。作为热交联剂,优选环氧化合物、至少具有2个烷氧基甲基或羟甲基的化合物。通过具有至少2个上述基团,与树脂及同种分子进行缩合反应形成交联结构体,可以使加热处理后的固化膜的机械强度、耐药品性提高。作为用作热交联剂的环氧化合物的优选例,例如,可举出双酚A型环氧树脂、双酚F型环氧树脂、丙二醇二缩水甘油醚、聚丙二醇二缩水甘油醚、聚甲基(缩水甘油基氧基丙基)硅氧烷等含有环氧基的聚硅氧烷等,但不限于此。具体而言,可举出EPICLON850-S、EPICLONHP-4032、EPICLONHP-7200、EPICLONHP-820、EPICLONHP-4700、EPICLONEXA-4710、EPICLONHP-4770、EPICLONEXA-859CRP、EPICLONEXA-1514、EPICLONEXA-4880、EPICLONEXA-4850-150、EPICLONEXA-4850-1000、EPICLONEXA-4816、EPICLONEXA-4822(以上商品名,大日本油墨化学工业(株)制)、RikaresinBEO-60E、RikaresinBPO-20E、RikaresinHBE-100、RikaresinDME-100(以上商品名,新日本理化(株)制)、EP-4003S,EP-4000S(以上商品名,AdekaCorporation制)、PG-100、CG-500、EG-200(以上商品名,OsakaGasChemicalsCo.,Ltd.制)、NC-3000、NC-6000(以上商品名,日本化药(株)制)、EPOX-MKR508、EPOX-MKR540、EPOX-MKR710、EPOX-MKR1710、VG3101L、VG3101M80(以上商品名,PrintechCo.,Ltd.制)、Celoxide2021P、Celoxide2081、Celoxide2083、Celoxide2085(以上商品名,DaicelChemicalIndustries,Ltd.制)等。作为用作热交联剂的至少具有2个烷氧基甲基或羟甲基的化合物,例如,可举出DML-PC、DML-PEP、DML-OC、DML-OEP、DML-34X、DML-PTBP、DML-PCHP、DML-OCHP、DML-PFP、DML-PSBP、DML-POP、DML-MBOC、DML-MBPC、DML-MTrisPC、DML-BisOC-Z、DML-BisOCHP-Z、DML-BPC、DML-BisOC-P、DMOM-PC、DMOM-PTBP、DMOM-MBPC、TriML-P、TriML-35XL、TML-HQ、TML-BP、TML-pp-BPF、TML-BPE、TML-BPA、TML-BPAF、TML-BPAP、TMOM-BP、TMOM-BPE、TMOM-BPA、TMOM-BPAF、TMOM-BPAP、HML-TPPHBA、HML-TPHAP、HMOM-TPPHBA、HMOM-TPHAP(以上,商品名,本州化学工业(株)制)、NIKALAC(注册商标)MX-290、NIKALACMX-280、NIKALACMX-270、NIKALACMX-279、NIKALACMW-100LM、NIKALACMX-750LM(以上,商品名,SanwaChemicalCo.,Ltd.制)。上述物质可以含有两种以上。热交联剂相对于聚酰亚胺前体树脂100重量份而言优选含有0.01~50重量份。聚酰亚胺前体树脂组合物可以含有无机填料及/或有机无机填料(organo-inorganicfiller)。作为无机填料,可举出二氧化硅微粒、氧化铝微粒、二氧化钛微粒、氧化锆微粒等。无机填料的形状没有特别限定,可举出球状、椭圆形状、扁平状、杆状、纤维状等。为了防止光的散射,所含有的无机填料优选粒径较小。平均粒径为0.5~100nm,优选为0.5~30nm的范围。无机填料的含量相对于聚酰亚胺前体而言,优选为1~200重量%,下限较优选为10重量%以上。上限较优选为150重量%以下,更优选为100重量%以下,特别优选为50重量%以下。随着含量的增加,柔性、耐折性降低。作为混合聚酰亚胺前体树脂组合物和无机填料的方法,可以使用各种已知方法。例如,可举出将无机填料、有机无机填料溶胶与聚酰亚胺前体进行混合。有机无机填料溶胶是指在有机溶剂中以30重量%左右的比例分散无机填料而得的物质,作为有机溶剂,可举出甲醇、异丙醇、正丁醇、乙二醇、甲基乙基酮、甲基异丁基酮、丙二醇单甲基乙酸酯(propyleneglycolmonomethylacetate)、丙二醇单甲基醚、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、1,3-二甲基咪唑啉酮、γ-丁内酯(gamma-butyllactone)等。就有机无机填料溶胶而言,通过使用硅烷偶联剂进行表面处理,无机填料在聚酰亚胺前体中的分散性提高。若硅烷偶联剂的末端官能团含有环氧基、氨基,则聚酰胺酸的羧酸与环氧基、氨基发生键合,由此与聚酰亚胺前体及、固化处理后的聚酰亚胺的亲和性提高,可以进行更高效的分散。作为具有环氧基的硅烷偶联剂,可举出2-(3,4-环氧基环己基)乙基三甲氧基硅烷、3-环氧丙氧基丙基甲基二甲氧基硅烷、3-环氧丙氧基丙基三甲氧基硅烷、3-环氧丙氧基丙基甲基二乙氧基硅烷、3-环氧丙氧基丙基三乙氧基硅烷等。作为具有氨基的硅烷偶联剂,可举出N-2-(氨基乙基)-3-氨基丙基甲基二甲氧基硅烷、N-2-(氨基乙基)-3-氨基丙基三甲氧基硅烷、3-氨基丙基三甲氧基硅烷、3-氨基丙基三乙氧基硅烷、3-三乙氧基甲硅烷基-N-(1,3-二甲基-亚丁基)丙基胺、N-苯基-3-氨基丙基三甲氧基硅烷等。作为有机无机填料溶胶的利用硅烷偶联剂进行的处理方法,可以使用各种已知方法。例如,可以在调整了浓度后的有机无机填料溶胶中添加硅烷偶联剂,于室温~80℃搅拌0.5~2小时,由此进行处理。聚酰亚胺前体树脂组合物可以含有光酸产生剂。通过含有光酸产生剂,在介由划出了曝光图案的掩模来照射光时,在曝光部产生酸,曝光部在碱性水溶液中的溶解性增大。如上所述含有光酸产生剂的聚酰亚胺前体树脂组合物可以用作正型感光性树脂(以下,有时也将含有光酸产生剂的聚酰亚胺前体树脂组合物称作正型感光性树脂组合物)。作为上述情况下使用的光酸产生剂,可举出醌二叠氮化合物、锍盐、鏻盐、重氮鎓盐、碘鎓盐等。其中从实现优异的溶解抑止效果、得到高灵敏度并且低膜损耗(lowfilmloss)的正型感光性树脂组合物的方面考虑,优选使用醌二叠氮化合物。另外,可以含有两种以上的光酸产生剂。由此,可以进一步增大曝光部与未曝光部的溶解速度之比,能够得到高灵敏度的正型感光性树脂组合物。作为醌二叠氮化合物,可举出在多羟基化合物中以酯的形式键合醌二叠氮的磺酸而得的化合物、在多氨基化合物中以磺酰胺的形式键合醌二叠氮的磺酸而得的化合物、在多羟基多氨基化合物中以酯及/或磺酰胺的形式键合醌二叠氮的磺酸而得的化合物等。所述多羟基化合物、多氨基化合物中的全部官能团不必均被醌二叠氮取代,但优选全部官能团中的50摩尔%以上被醌二叠氮取代。通过使用上述醌二叠氮化合物,能够得到通过通常的紫外线即汞灯的i线(波长365nm)、h线(波长405nm)、g线(波长436nm)进行反应的正型感光性树脂组合物。在上述情况下,醌二叠氮化合物优选使用5-萘醌二叠氮磺酰基、4-萘醌二叠氮磺酰基中的任一种。可以使用在同一分子中具有上述基团中的两者的化合物,也可以并用使用了不同基团的化合物。上述情况下所使用的醌二叠氮化合物可以由特定的酚类化合物通过以下方法进行合成。例如可举出使5-萘醌二叠氮磺酰氯与酚类化合物在三乙基胺存在下进行反应的方法。酚类化合物的合成方法可举出在酸催化剂下使α-(羟基苯基)苯乙烯衍生物与多元酚化合物反应的方法等。上述情况下所使用的光酸产生剂的含量相对于聚酰亚胺前体100重量份而言优选为3~40重量份。通过使光酸产生剂的含量在该范围,能够实现更高灵敏度化。也可以根据需要进一步含有敏化剂等。形成正型感光性树脂组合物的图案时,将正型感光性树脂组合物的清漆涂布在支承基板上,曝光后,使用显影液除去曝光部。作为显影液,优选四甲基氢氧化铵、二乙醇胺、二乙基氨基乙醇、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、三乙基胺、二乙基胺、甲胺、二甲基胺、乙酸二甲基氨基乙基酯、二甲基氨基乙醇、甲基丙烯酸二甲基氨基乙基酯、环己基胺、乙二胺、六亚甲基二胺等显示碱性的化合物的水溶液。另外,根据情况,可以在上述碱性水溶液中单独添加或者组合添加多种下述物质:N-甲基-2-吡咯烷酮、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲基亚砜、γ-丁内酯、二甲基丙烯酰胺等极性溶剂、甲醇、乙醇、异丙醇等醇类、乳酸乙酯、丙二醇单甲基醚乙酸酯等酯类、环戊酮、环己酮、异丁酮、甲基异丁基酮等酮类等。优选在显影后利用水进行冲洗处理。此处,也可以将乙醇、异丙基醇等醇类、乳酸乙酯、丙二醇单甲基醚乙酸酯等酯类等加入到水中进行冲洗处理。<聚酰亚胺树脂膜>以下说明本发明的聚酰亚胺前体的制造方法。聚酰胺酸、聚酰胺酸酯、聚酰胺酸甲硅烷基酯等聚酰亚胺前体可以通过二胺化合物与酸二酐或其衍生物的反应进行合成。作为衍生物,可举出该酸二酐的四甲酸、酰氯化物、四甲酸的单、二、三或四酯等,具体可举出利用甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基等进行酯化而得的结构。就聚合反应的反应方法而言,只要可制造目标聚酰亚胺前体则没有特别限定,可以使用已知的反应方法。作为具体的反应方法,可举出将规定量的全部二胺成分及反应溶剂加入到反应器中使其溶解后,加入规定量的酸二酐成分,于室温~80℃搅拌0.5~30小时的方法等。以下,对使用含有本发明的聚酰亚胺前体和溶剂的聚酰亚胺前体树脂组合物制造聚酰亚胺树脂膜的方法进行说明。需要说明的是,聚酰亚胺树脂膜中也可以含有上述表面活性剂、内部脱模剂、硅烷偶联剂、热交联剂、无机填料、光酸产生剂等。首先,将聚酰亚胺前体树脂组合物涂布在支承基板上形成聚酰亚胺前体树脂组合物膜。作为支承基板,例如可使用硅、陶瓷类、砷化镓、钠钙玻璃、无碱玻璃等,但不限于此。涂布方法例如有狭缝涂布法、旋涂法、喷涂法、辊涂法、棒涂法等方法,也可以组合上述方法进行涂布。上述方法中,优选通过旋涂法或者狭缝涂布法进行涂布。接着,将涂布于支承基板上的聚酰亚胺前体树脂组合物进行干燥,得到聚酰亚胺前体树脂膜。干燥使用加热板、烘箱、红外线、真空箱等。使用加热板时,在板上直接保持支承基板(其涂布有聚酰亚胺前体树脂组合物)进行加热,或者在设置于板上的固定销(proxypins)等夹具上保持支承基板(其涂布有聚酰亚胺前体树脂组合物)进行加热。作为固定销的材质,有铝、不锈钢等金属材料、或者聚酰亚胺树脂、“Teflon(注册商标)”等合成树脂,可以使用任意材质的固定销。固定销的高度根据支承基板的尺寸、作为被加热体的树脂层的种类、加热目的等而有所不同,例如对涂布于300mm×350mm×0.7mm的玻璃支承基板上的树脂层进行加热时,固定销的高度优选为2~12mm左右。其中,优选使用真空箱进行真空干燥,更优选在真空干燥后进一步进行用于干燥的加热,或者一边进行真空干燥一边进行用于干燥的加热。由此,可以实现干燥处理时间的缩短、及均匀的涂布膜的形成。用于干燥的加热温度根据支承基板、聚酰亚胺前体的种类、目的而有所不同,优选在室温~170℃的范围进行1分钟~数小时。进而,干燥工序可以在相同条件、或不同条件下进行多次。接着,进行用于酰亚胺化的加热。将聚酰亚胺前体树脂膜在180℃以上400℃以下的范围进行加热而将其转化为聚酰亚胺树脂膜。需要说明的是,就热酰亚胺化工序而言,可以在上述干燥工序之后经过任何工序后进行。热酰亚胺化工序的气氛没有特别限定,可以为空气,也可以为氮、氩等非活性气体。其中,由于本发明的聚酰亚胺树脂膜要求无色透明性,所以优选在氧浓度为5%以下的气氛下加热进行热酰亚胺化。通常,通过降低氧浓度,可以减少加热时聚酰亚胺膜的着色,得到显示高透明性的聚酰亚胺树脂膜。另外,热酰亚胺化工序中,可以选择与生产线的烘箱的加热形式对应的升温方法,但是优选经5~120分钟升温至最高加热温度。例如,可以在烘箱内将形成在基材上的聚酰亚胺前体树脂膜经5~120分钟从室温升温至用于热酰亚胺化的加热温度从而进行酰亚胺化,制成聚酰亚胺树脂膜;也可以将在基材上形成的聚酰亚胺前体树脂膜直接投入预先加热至200℃以上650℃以下的范围的烘箱内,进行加热处理从而进行酰亚胺化,制成聚酰亚胺树脂膜。将如上所述在支承基板上形成的聚酰亚胺树脂膜从支承基板上剥离时,可举出机械剥离的方法;浸渍于氢氟酸等药液、水中的方法;对固化膜与支承基板的界面照射激光的方法等,但是可以使用任意方法。如上所述得到的聚酰亚胺树脂膜具有高透明性、低双折射、低CTE、高耐热性、耐药品性、柔性,可以作为挠性基板适合地用于后述的液晶显示器、有机EL显示器、电子纸等显示元件、滤色片、光波导等光学元件、太阳能电池、CMOS等受光元件、触摸面板、电路基板等。聚酰亚胺树脂膜的透明性可以通过测定紫外-可见光(UV-vis)光谱进行判断。作为透明性树脂膜,就厚度为10μm的膜而言,波长400nm处的光线透过率优选为70%以上,较优选为80%以上,更优选为90%以上。就本发明的聚酰亚胺树脂膜而言,以相对于膜表面为垂直方向与相对于膜表面为水平方向的折射率差定义的双折射优选为0.06以下。另外,就线性热膨胀系数(CTE)而言,优选50℃~200℃的平均值为40ppm/℃以下。较优选双折射为0.04以下并且CTE为40ppm/℃以下,更优选双折射为0.04以下并且CTE为30ppm/℃以下。通常,在支承基板上成膜的聚酰亚胺树脂膜中产生残留应力,其结果,支承基板发生翘曲。支承基板的翘曲引起元件的加工精度的降低、元件加工时的搬送失误等不良情况。聚酰亚胺树脂膜的残留应力主要是由于支承基板与聚酰亚胺树脂膜的CTE不同而引起的,若两者的CTE之差增大,则残留应力也增大。通常,支承基板使用与聚酰亚胺树脂膜相比CTE较低的玻璃基板等,因此聚酰亚胺树脂膜要求低CTE。另一方面,显示器基板等显示元件、滤色片等光学元件的基板要求低双折射。例如,对于利用使用了双折射高的基板的显示元件、滤色片而成的显示器而言,产生图像失真、斜视野(obliquevisualfields)中的颜色漂移等不良情况。本发明的聚酰亚胺树脂膜的低CTE和低双折射的均衡性优异,因此可以同时实现高加工精度和高品质的显示图像。关于耐药品性,由本发明的聚酰亚胺前体形成的聚酰亚胺对于丙酮、丙二醇单甲基醚、N-甲基-2-吡咯烷酮、γ-丁内酯、二甲基亚砜等有机溶剂、盐酸、硝酸、磷酸、乙酸、草酸等酸性溶液、四甲基氢氧化铵等碱性溶液显示优异的耐药品性。需要说明的是,就上述物性值而言,在聚酰亚胺树脂膜中含有表面活性剂、内部脱模剂、硅烷偶联剂、热交联剂、无机填料、光酸产生剂等时,为作为也含有上述物质的状态的树脂膜的物性值。本发明的聚酰亚胺树脂膜可以用于液晶显示器、有机EL显示器、电子纸等显示元件、滤色片、光波导等光学元件、太阳能电池、CMOS等受光元件、触摸面板、电路基板等。特别是将上述显示元件、受光元件等活用作具有柔软性可显著弯曲、或可折弯的挠性元件时,可以优选使用本发明的聚酰亚胺树脂膜作为挠性基板。需要说明的是,关于将本发明的聚酰亚胺树脂膜用作挠性基板时的显示元件、光学元件(滤色片等)等,如挠性显示元件、挠性光学元件(挠性滤色片等)等那样,也有时在元件名之前标记“挠性”进行表示。就显示元件、受光元件、电路、TFT基板等的制造而言,可以在支承基板上形成聚酰亚胺树脂膜,将该聚酰亚胺树脂膜从支承基板上剥离,然后在聚酰亚胺树脂膜上实施,也可以不从支承基板上剥离,直接在聚酰亚胺树脂膜上实施。后者的情况下,在制造显示元件、受光元件、电路、TFT基板等后,从支承基板上将其剥离。该方法具有可以利用现有的单张式的制造工艺的优点。另外,因为聚酰亚胺树脂膜被固定于支承基板,所以适合用于位置精度良好地制造显示元件、受光元件、电路、TFT基板、触摸面板等。以下说明中多以后者的方法作为代表例进行说明,但也可以均为前者的方法。将形成显示元件、受光元件、电路、TFT基板、触摸面板等的聚酰亚胺树脂膜从基材上剥离的方法没有特别限定。作为剥离方法的例子,可举出浸渍在水中的方法;浸渍在盐酸、氢氟酸等药液中的方法;对聚酰亚胺树脂膜与支承基板的界面照射紫外部光线至红外部光线的波长范围的激光的方法等。需要说明的是,为了容易剥离,可以在将聚酰亚胺前体树脂组合物向基材涂布之前,在支承基板上预先涂布脱模剂,或者预先制备牺牲层。作为脱模剂,可举出植物油系、醇酸系、聚硅氧烷系、氟系、芳香族高分子系、烷氧基硅烷系等。作为牺牲层,可举出金属膜、金属氧化物膜、无定形硅膜等。可以在本发明的聚酰亚胺树脂膜的至少一方的面上制备无机膜作为气体阻隔层,可以以带有气体阻隔层的基板的形式,适合地用于显示元件的基板。聚酰亚胺树脂膜上的气体阻隔层起到防止水蒸气、氧等透过的作用。特别是就有机电致发光元件(有机EL元件)而言,由于因水分导致的元件劣化比较明显,所以需要对基板赋予气体阻隔性。<挠性基板>含有本发明的聚酰亚胺树脂膜的基板具有柔软性,具有能够显著弯曲的优点。将所述具有柔软性的基板称作挠性基板。挠性基板可以经过至少以下工序(1)~工序(3)进行制造。另外,在聚酰亚胺树脂膜上具有无机膜的挠性基板可以经过至少以下(1)~(4)工序进行制造。工序(1),将含有本发明的聚酰亚胺前体和溶剂的聚酰亚胺前体树脂组合物涂布在支承基板上;工序(2),从涂布后的聚酰亚胺前体树脂组合物中除去溶剂;工序(3),将聚酰亚胺前体进行酰亚胺化得到聚酰亚胺树脂膜;工序(4),在聚酰亚胺树脂膜上形成无机膜。对于上述工序(1)~工序(3),如在<聚酰亚胺树脂膜>中已经详细说明过的那样。上述工序(4)是在聚酰亚胺树脂膜的至少一面形成无机膜的工序。可以将聚酰亚胺树脂膜从支承基板上剥离,制造挠性基板。需要说明的是,就工序(4)而言,可以在聚酰亚胺树脂膜上直接形成无机膜,也可以中间介由其他层而形成无机膜。优选在聚酰亚胺树脂膜上直接形成无机膜的方法。就制造挠性基板时的支承基板而言,优选具有自立性(self-standable)的、硬质的、并且涂布树脂组合物的面平滑、具有耐热性的基板。材质没有特别限定,例如可举出钠玻璃、无碱玻璃、硅、石英、氧化铝、蓝宝石等陶瓷、砷化镓、铁、锡、锌、铜、铝、不锈钢等金属、聚酰亚胺、聚苯并噁唑等耐热塑料膜、聚四氟乙烯、聚偏氟乙烯等氟树脂、环氧树脂、聚对苯二甲酸乙二醇酯、聚萘二甲酸乙二醇酯等基材。上述物质中,从表面平滑性、可进行激光剥离的方面、廉价的方面等考虑,优选玻璃。玻璃的种类没有特别限定,从降低金属杂质的观点考虑,优选无碱玻璃。如上所述显示元件的基板使用挠性基板的情况下,由于基板要求气体阻隔性,所以优选在聚酰亚胺树脂膜上形成无机膜。作为构成无机膜(其用作气体阻隔层)的材料,可优选使用金属氧化物、金属氮化物及金属氮氧化物。例如,可举出铝(Al)、硅(Si)、钛(Ti)、锡(Sn)、锌(Zn)、锆(Zr)、铟(In)、铌(Nb)、钼(Mo)、钽(Ta)、钙(Ca)等金属的氧化物、金属氮化物及金属氮氧化物。对于至少含有Zn、Sn、In的金属氧化物、金属氮化物及金属氮氧化物的气体阻隔层而言,耐弯曲性高,特别优选。对于Zn、Sn、In的原子浓度为20~40%的气体阻隔层而言,耐弯曲性更高,更优选。气体阻隔层中共存有二氧化硅、氧化铝的组成的耐弯曲性也良好,为优选。所述无机气体阻隔层例如可通过溅射法、真空蒸镀法、离子镀法、等离子体CVD法等从气相中沉积材料形成膜的气相沉积法进行制作。其中,就溅射法而言,通过进行反应性溅射(在含氧气氛下溅射金属靶)能够提高制膜速度。气体阻隔层的形成可以在包含支承基板和聚酰亚胺树脂膜的层合体上进行,也可以在已经从支承基板上剥离的自立膜上进行。本发明的聚酰亚胺树脂的耐热性高,所以可以升高基板温度制作气体阻隔层。气体阻隔层的制膜温度优选为80~400℃。为了提高气体阻隔性能,选择高制膜温度是有利的。若制膜温度高则有时耐弯曲性降低,因此耐弯曲性为重要性能的情况下,优选气体阻隔层的制膜温度为100~300℃。本发明的聚酰亚胺树脂膜由于具有优异的耐热性,所以即使在高温下(例如300℃)形成气体阻隔层,也不会在膜中产生褶皱等缺陷。气体阻隔层的层数没有限定,可以仅为1层,也可以为2层以上的多层。作为多层膜的例子,可举出包含第1层为SiO、第2层为SiN的气体阻隔层;包含第1层为SiO/AlO/ZnO、第2层为SiO的气体阻隔层。在挠性基板的气体阻隔层上形成有机EL发光层等具有各种功能的层、制作显示元件、光学元件等的工序中,可以使用各种有机溶剂。例如,就滤色片(以下,记作“CF”)而言,在聚酰亚胺树脂膜上形成气体阻隔层,然后形成着色像素、黑矩阵等制成CF。此时,气体阻隔层的耐溶剂性差时,气体阻隔性能降低。因此,优选对最上层的气体阻隔层赋予耐溶剂性,例如最上层的气体阻隔层优选包含氧化硅。气体阻隔层的组成分析,可以通过使用X射线光电子能谱法(XPS法)对各元素进行定量分析来进行。气体阻隔层的总厚度优选为20~600nm,更优选为30~300nm。气体阻隔层的厚度通常可以通过利用透射电子显微镜(TEM)进行截面观察来测定。由于气体阻隔层的上层与下层的边界区域的组成以沿着倾斜方向的方式变化等原因而导致利用TEM无法目视识别明确的界面时,首先,进行厚度方向的组成分析,求出厚度方向的元素的浓度分布,然后基于浓度分布的信息求出层的边界及层的厚度。厚度方向的组成分析的顺序及各层的层的边界以及层厚度的定义记载如下。首先,利用透射电子显微镜观察气体阻隔层的截面,测定整体的厚度。接着,应用可进行深度方向上元素的组成分析的下述测定,得到与气体阻隔层的厚度位置对应的元素的浓度分布(厚度方向的浓度概况(profile))。作为此时适用的组成分析方法,可举出电子能量损失光谱法(以下记作EELS分析)、能量色散型X射线光谱法(以下记作EDX分析)、二次离子质谱法(以下记作SIMS分析)、X射线光电子能谱法(记作XPS分析)、俄歇电子能谱法(以下记作AES分析),但从灵敏度及精度的观点考虑,更优选EELS分析。因此,首先进行EELS分析,以后按照之前列举的顺序(EELS分析→EDX分析→SIMS分析→XPS分析→AES分析)进行分析,对于利用较上位的分析无法鉴定的成分,应用下位的分析数据。<CF>在使用了本发明的聚酰亚胺树脂膜的挠性基板上设置黑矩阵、着色像素,由此得到CF。该CF将树脂膜用于基材,因此具有轻质、不易破裂、柔性等特征。优选黑矩阵、着色像素层中的至少1层中使用的树脂含有聚酰亚胺树脂。进而,从减小反射率及耐热性的观点考虑,优选黑矩阵包含低光学浓度层、和形成于该低光学浓度层上的高光学浓度层,并且低光学浓度层和高光学浓度层的至少1层中使用的树脂含有聚酰亚胺树脂。本发明的聚酰亚胺树脂膜对于作为聚酰亚胺前体的溶剂的通常的极性非质子性溶剂具有高耐药品性,因此可以在黑矩阵、着色像素层中使用聚酰亚胺树脂。进而,在黑矩阵、着色像素层上形成气体阻隔层的情况下,黑矩阵、着色像素层的聚酰亚胺树脂的耐热性高,因此在气体阻隔层的形成过程中较少产生气体,对于气体阻隔性高的气体阻隔层的制膜而言有利。另外,在黑矩阵、着色像素层的图案加工时,由于可以作为可溶于碱性水溶液的聚酰亚胺前体使用,所以对于微细图案的形成而言有利。通过附图说明CF的构成例。图1A表示在支承基板上形成的、包含本发明的聚酰亚胺树脂膜的CF的基本构成。利用上述剥离方法从其中剥离支承基板1,由此得到以本发明的聚酰亚胺树脂膜作为基板的CF。在支承基板1上形成聚酰亚胺树脂膜2,在其上形成黑矩阵4、红色的着色像素5R、绿色的着色像素5G及蓝色的着色像素5B,进而具备保护层(overcoatlayer)6。需要说明的是,保护层6并非必需,不配置保护层6的例子也是可以的。另外,图1B~图1D为图1A的变形例,进一步形成有作为无机膜的气体阻隔层3。形成气体阻隔层3时,没有特别限定,例如,可以在聚酰亚胺树脂膜2上形成(参见图1B);也可以在黑矩阵4、着色像素层上形成(参见图1C);也可以在存在于滤色片表面的保护层6上形成;也可以在聚酰亚胺树脂膜2上和保护层6上这两者上形成(参见图1D)。另外,气体阻隔层的层数没有限定,可以仅为1层,也可以为2层以上的多层。作为多层膜的例子,可举出包含第1层为SiO、第2层为SiN的气体阻隔层;包含第1层为SiO/AlO/ZnO、第2层为SiO的气体阻隔层。(黑矩阵)黑矩阵优选为由将黑色颜料分散在树脂中而得的树脂形成的黑矩阵。作为黑色颜料的例子,可举出炭黑、钛黑、氧化钛、氧氮化钛、氮化钛或四氧化三铁。特别优选炭黑、钛黑。另外,也可以混合红颜料、绿颜料、蓝颜料制成黑色颜料使用。作为用于树脂黑矩阵的树脂,从耐热性的观点、形成微细图案的容易程度的观点考虑,优选聚酰亚胺树脂。聚酰亚胺树脂优选为将由酸二酐和二胺合成得到的聚酰胺酸进行图案加工、然后进行热固化而制成的聚酰亚胺树脂。作为酸二酐、二胺及溶剂的例子,可以使用在上述聚酰亚胺树脂中列举的物质。作为用于树脂黑矩阵的树脂,也可使用感光性丙烯酸树脂。包含分散有黑色颜料的、碱可溶性的丙烯酸树脂、光聚合性单体、高分子分散剂及添加剂。作为碱可溶性的丙烯酸树脂的例子,可举出不饱和羧酸与烯键式不饱和化合物形成的共聚物。作为不饱和羧酸的例子,可举出丙烯酸、甲基丙烯酸、衣康酸、丁烯酸、马来酸、富马酸、乙烯基乙酸或酸酐。作为光聚合性单体的例子,可举出三羟甲基丙烷三(甲基)丙烯酸酯、季戊四醇三(甲基)丙烯酸酯、1,3,5-三丙烯酰基六氢-1,3,5-三嗪(triacrylformal)、季戊四醇四(甲基)丙烯酸酯、二季戊四醇六(甲基)丙烯酸酯或二季戊四醇五(甲基)丙烯酸酯。作为光聚合引发剂的例子,可举出二苯甲酮、N,N’-四乙基-4,4’-二氨基二苯甲酮、4-甲氧基-4’-二甲基氨基二苯甲酮、2,2-二乙氧基苯乙酮、α-羟基异丁基苯酮(α-hydroxyisobutylphenon)、噻吨酮或2-氯噻吨酮。作为用于溶解感光性丙烯酸树脂的溶剂的例子,可举出丙二醇单甲基醚乙酸酯、丙二醇单乙基醚乙酸酯、乙酰乙酸乙酯、甲基-3-甲氧基丙酸酯、乙基-3-乙氧基丙酸酯、甲氧基丁基乙酸酯或3-甲基-3-甲氧基丁基乙酸酯。为了抑制由外部光线反射引起的目视识别性的降低,黑矩阵优选为由低光学浓度层、和形成在该低光学浓度层上的高光学浓度层形成的层合树脂黑矩阵。需要说明的是,所谓低光学浓度层,是指光学浓度并非0、且实质上不透明的层结构,其每单位厚度的光学浓度的值小于高光学浓度层的每单位厚度的光学浓度。构成上述层合树脂黑矩阵的树脂没有特别限定,从将低光学浓度层和高光学浓度层一并地进行图案形成的观点考虑,优选低光学浓度层为聚酰亚胺树脂,高光学浓度层为丙烯酸树脂。进而,为了降低反射率,较优选上述树脂黑矩阵中包含微粒。(着色像素)形成黑矩阵后,形成着色像素。着色像素包含红、绿、蓝3色的着色像素。另外,除了3色的着色像素之外,还可以形成无色透明或极薄地着色的第4色的像素,由此也能够提高显示装置的白色显示的明亮度。滤色片的着色像素可以使用含有颜料或染料作为着色剂的树脂。作为用于红色着色像素的颜料的例子,可举出PR254、PR149、PR166、PR177、PR209、PY138、PY150或PYP139,作为用于绿色着色像素的颜料的例子,可举出PG7、PG36、PG58、PG37、PB16、PY129、PY138、PY139、PY150或PY185,作为用于蓝色着色像素的颜料的例子,可举出PB15:6或PV23。作为蓝色染料的例子,可举出C.I.BasicBlue(BB)5、BB7、BB9或BB26,作为红色染料的例子,可举出C.I.AcidRed(AR)51、AR87或AR289,作为绿色染料的例子,可举出C.I.AcidGreen(AG)25、AG27。作为用于红绿蓝色着色像素的树脂的例子,可举出丙烯酸系树脂、环氧系树脂或聚酰亚胺系树脂。从耐热性的观点考虑,优选聚酰亚胺树脂,为了降低CF的制造成本,可以使用感光性丙烯酸系树脂。为了形成包含聚酰亚胺树脂的着色像素,通常的方法如下:将至少包含聚酰胺酸、着色剂、溶剂的非感光性彩色糊剂涂布在基板上后,通过风干、加热干燥、真空干燥等进行干燥,形成非感光性聚酰胺酸着色被膜,使用正型光致抗蚀剂形成所期望图案后,将光致抗蚀剂进行碱剥离,最后于200~300℃加热1分钟~3小时,由此使着色像素固化(聚酰亚胺化)。感光性丙烯酸系树脂通常含有碱可溶性的丙烯酸树脂、光聚合性单体及光聚合引发剂。作为碱可溶性的丙烯酸树脂的例子,可举出不饱和羧酸与烯键式不饱和化合物形成的共聚物。作为不饱和羧酸的例子,可举出丙烯酸、甲基丙烯酸、衣康酸、丁烯酸、马来酸、富马酸、乙烯基乙酸或酸酐。作为光聚合性单体的例子,可举出三羟甲基丙烷三(甲基)丙烯酸酯、季戊四醇三(甲基)丙烯酸酯、1,3,5-三丙烯酰基六氢-1,3,5-三嗪、季戊四醇四(甲基)丙烯酸酯、二季戊四醇六(甲基)丙烯酸酯或二季戊四醇五(甲基)丙烯酸酯。作为光聚合引发剂的例子,可举出二苯甲酮、N,N’-四乙基-4,4’-二氨基二苯甲酮、4-甲氧基-4’-二甲基氨基二苯甲酮、2,2-二乙氧基苯乙酮、α-羟基异丁基苯酮、噻吨酮或2-氯噻吨酮。作为用于溶解感光性丙烯酸系树脂的溶剂的例子,可举出丙二醇单甲基醚乙酸酯、丙二醇单乙基醚乙酸酯、乙酰乙酸乙酯、甲基-3-甲氧基丙酸酯、乙基-3-乙氧基丙酸酯、甲氧基丁基乙酸酯或3-甲基-3-甲氧基丁基乙酸酯。为了使形成有黑矩阵及着色像素的滤色片的表面平坦化,可以在滤色片表面进一步形成保护层。作为用于形成保护层的树脂的例子,可举出环氧树脂、丙烯酸环氧树脂、丙烯酸树脂、硅氧烷树脂或聚酰亚胺树脂。作为保护层的厚度,优选表面变平坦的厚度,较优选0.5~5.0μm,更优选1.0~3.0μm。(CF的制造方法)含有本发明的聚酰亚胺树脂膜的CF可以经过至少以下工序进行制造。工序(1),将含有本发明的聚酰亚胺前体和溶剂的聚酰亚胺前体树脂组合物涂布在支承基板上;工序(2),从涂布后的聚酰亚胺前体树脂组合物中除去溶剂;工序(3),将聚酰亚胺前体进行酰亚胺化得到聚酰亚胺树脂膜;工序(4),在聚酰亚胺树脂膜上形成黑矩阵及着色像素;工序(5),从支承基板上剥离聚酰亚胺树脂膜。上述工序(1)~工序(3)如在<聚酰亚胺树脂膜>中已经详细说明过的那样。上述工序(4)是在聚酰亚胺树脂膜上形成黑矩阵及着色像素的工序。如下所述,黑矩阵、着色像素的图案形成中使用光刻法。目前,作为液晶显示器、有机EL显示器,要求300ppi以上的高清晰度,挠性显示器面板也要求同等以上的性能。为了实现上述高分辨率,必需进行高精度的图案形成。在聚酰亚胺树脂膜(其是在支承基板上制膜得到的)上形成黑矩阵等制作CF时,可以应用作为支承基板使用玻璃基板并在其上形成黑矩阵等制作CF的现行技术,与在聚酰亚胺自立膜上制作CF的情况相比,在形成高清晰度图案方面有利。需要说明的是,对于工序(4)而言,可以在聚酰亚胺树脂膜上直接形成黑矩阵、着色像素,也可以在中间介由其他层形成黑矩阵、着色像素。在上述CF的制造工序中,还可以包括形成气体阻隔层等无机膜的工序。形成无机膜的位置没有特别限定。例如,可以形成在聚酰亚胺树脂膜上,可以形成在黑矩阵、着色像素层上,也可以形成在存在于滤色片表面的平坦化膜上,还可以形成在聚酰亚胺树脂膜上和平坦化膜上这两者上。另外,无机膜的层数没有限定,可以仅为1层,也可以为2层以上的多层。作为多层膜的例子,可举出包含第1层为SiO、第2层为SiN的无机膜;包含第1层为SiO/AlO/ZnO、第2层为SiO的无机膜。接着,更具体地说明本发明的CF的制造方法之一例。通过上述方法在支承基板上制作聚酰亚胺树脂膜及气体阻隔层。在其上通过利用旋涂机或模涂机等的方法以固化后的厚度成为1μm的方式涂布包含聚酰胺酸(其分散有包含炭黑或钛黑的黑色颜料)的黑矩阵用糊剂,减压干燥直至60Pa以下,然后利用110~140℃的热风烘箱或加热板进行半固化。通过利用旋涂机或模涂机等的方法涂布正型抗蚀剂以使预焙后的厚度成为1.2μm,然后,进行减压干燥直至80Pa,利用80~110℃的热风烘箱或加热板进行预焙,形成抗蚀剂膜。之后,利用接近式曝光机(proximityexposureapparatus)或投影曝光机(projectionexposureapparatus)等介由光掩模利用紫外线选择地进行曝光,之后在1.5~3重量%的氢氧化钾或四甲基氢氧化铵等碱显影液中浸渍20~300秒,由此除去曝光部。使用剥离液剥离正型抗蚀剂后,利用200~300℃的热风烘箱或加热板加热10~60分钟,使聚酰胺酸转化为聚酰亚胺,由此形成树脂黑矩阵。着色像素使用着色剂和树脂进行制作。使用颜料作为着色剂时,在颜料中混合高分子分散剂及溶剂进行分散处理,在得到的分散液中添加聚酰胺酸而进行制作。另一方面,使用染料作为着色剂时,在染料中添加溶剂、聚酰胺酸进行制作。此时的全部固态成分为作为树脂成分的高分子分散剂、聚酰胺酸、与着色剂的总量。通过利用旋涂机或模涂机等的方法将得到的着色剂组合物涂布在形成有树脂黑矩阵的聚酰亚胺树脂膜上,以使加热处理后的厚度成为0.8~3.0μm的目标厚度,然后进行减压干燥直至80Pa,利用80~110℃的热风烘箱或加热板进行预焙,形成着色剂的涂膜。接着,通过利用旋涂机或模涂机等的方法涂布正型抗蚀剂以使预焙后的厚度成为1.2μm,然后,进行减压干燥直至80Pa,利用80~110℃的热风烘箱或加热板进行预焙,形成抗蚀剂膜。之后,利用接近式曝光机或投影曝光机等介由光掩模利用紫外线选择地进行曝光,然后浸渍在1.5~3重量%的氢氧化钾或四甲基氢氧化铵等碱显影液中20~300秒,由此除去曝光部。使用剥离液剥离正型抗蚀剂后,利用200~300℃的热风烘箱或加热板加热10~60分钟,由此使聚酰胺酸转化为聚酰亚胺,从而形成着色像素。使用针对不同颜色的着色像素而制作的着色剂组合物,针对红色着色像素、绿色着色像素及蓝色着色像素依次进行上述图案形成工序。需要说明的是,着色像素的图案形成的顺序没有特别限定。之后,通过利用旋涂机或模涂机等的方法涂布聚硅氧烷树脂后,进行真空干燥,利用80~110℃的热风烘箱或加热板进行预焙,利用150~250℃的热风烘箱或加热板加热5~40分钟,由此形成保护层,从而可以制作本发明的CF的像素。如上所述,本发明的聚酰亚胺树脂膜由于CTE低,所以在支承基板上形成聚酰亚胺树脂膜时可以减少基板翘曲。因此,可以减小在形成黑矩阵、着色像素时在光刻工序中的散焦,结果能够高精度地制作CF。<TFT基板>本发明的聚酰亚胺树脂膜可以适合地用于TFT基板的基材。即,可以得到在本发明的聚酰亚胺树脂膜上具备了TFT的TFT基板。所述TFT基板将树脂膜用于基材,因此具有轻质、不易破裂等特征。利用了本发明的聚酰亚胺树脂膜的TFT基板可以经过至少以下工序进行制造。工序(1),将含有本发明的聚酰亚胺前体和溶剂的聚酰亚胺前体树脂组合物涂布在支承基板上;工序(2),从涂布后的聚酰亚胺前体树脂组合物中除去溶剂;工序(3),将聚酰亚胺前体进行酰亚胺化得到聚酰亚胺树脂膜;工序(4),在聚酰亚胺树脂膜上形成气体阻隔层;工序(5),在聚酰亚胺树脂膜上形成TFT。对于上述工序(1)~工序(3)而言,如在<聚酰亚胺树脂膜>中已经详细说明的那样。上述工序(4)及工序(5)是在聚酰亚胺树脂膜上形成气体阻隔层,接着形成TFT的工序。需要说明的是,工序(4)、工序(5)可以在聚酰亚胺树脂膜上直接形成气体阻隔层、TFT,也可以在中间介由其他层形成气体阻隔层、TFT。优选在聚酰亚胺树脂膜上直接形成气体阻隔层、在其上形成TFT的方法。作为用于形成TFT的半导体层,可举出无定形硅半导体、多晶硅半导体、In-Ga-ZnO-4为代表的氧化物半导体、并五苯、聚噻吩为代表的有机物半导体。例如,使用本发明的聚酰亚胺树脂膜作为基材,通过已知方法依次形成气体阻隔膜、栅电极、栅绝缘膜、半导体层、蚀刻阻挡膜、源·漏极,制作底栅型TFT。经过上述工序可以制造利用了聚酰亚胺树脂膜的TFT基板。所述TFT基板可以用作液晶元件、有机EL元件、电子纸等显示元件的驱动基板。在TFT基板的制造中,在形成栅电极、栅绝缘膜、半导体层、蚀刻阻挡膜、源·漏极时主要采用光刻法。如上所述,本发明的聚酰亚胺树脂膜的CTE低,因此在支承基板上形成聚酰亚胺树脂膜时能够减少基板的翘曲。因此,能够减小在光刻工序中的散焦,因而能够以高精度制作TFT。结果,能够得到驱动性能良好的TFT基板。另外,例如在底部发光型有机EL显示器的情况下,显示器的使用者目视识别透过TFT基板的光。因此,若将双折射高的树脂用于TFT基板的基材,则从斜向(obliquedirection)观察时产生颜色漂移等。本发明的聚酰亚胺与现有的低CTE/透明聚酰亚胺相比双折射低,因此能够改善上述不良情况。<触摸面板>使用了本发明的聚酰亚胺树脂膜的挠性基板能够用于触摸面板的基板。例如,通过在本发明的聚酰亚胺树脂膜的至少一面形成透明导电层,由此制成透明导电膜,通过使用粘结剂、粘合剂等将透明导电膜彼此层合,从而可以制作触摸面板。作为透明导电层,可以应用已知的金属膜、金属氧化物膜等、碳纳米管、石墨烯等碳材料,其中从透明性、导电性及机械特性的观点考虑,优选应用金属氧化物膜。作为上述金属氧化物膜,可举出例如,作为杂质添加有锡、碲、镉、钼、钨、氟、锌、锗等的氧化铟、氧化镉及氧化锡、作为杂质添加有铝的氧化锌、氧化钛等金属氧化物膜。其中,含有2~15质量%氧化锡或氧化锌的氧化铟的薄膜由于透明性及导电性优异,故可以优选使用。上述透明导电层的成膜方法只要是能够形成目标薄膜的方法,则可以为任意方法,例如,适合采用溅射法、真空蒸镀法、离子镀法、等离子体CVD法等从气相中沉积材料而形成膜的气相沉积法等。其中,从能够得到优异的导电性·透明性的观点考虑,特别优选采用溅射法进行成膜。另外,透明导电层的膜厚优选为20~500nm,更优选为50~300nm。<电路基板>使用了本发明的聚酰亚胺树脂膜的挠性基板可以用于电路基板。作为电路基板没有特别限定,可举出将本发明的聚酰亚胺树脂膜作为基础膜,在其上形成任意电路的电路基板。例如,将本发明的聚酰亚胺树脂膜作为基础膜,在其一面或两面介由粘结剂层设置铜箔,在该覆盖有铜的聚酰亚胺膜(CCL)上形成光致抗蚀膜,进行曝光/显影、蚀刻、抗蚀剂剥离、阻焊膜形成、电解镀金,在其上粘贴用作保护层的覆盖层膜(coverlayfilm),由此可以得到电路基板。如上所述,因为本发明的聚酰亚胺树脂膜的透明性高,所以可以得到透明电路基板。其可以适合用于透明显示器。<显示元件、受光元件>利用了本发明的聚酰亚胺的挠性基板可以用于液晶显示器、有机EL显示器、电子纸之类的显示元件、太阳能电池、CMOS等受光元件。将所述显示元件、受光元件活用作可折弯的挠性装置时,可以特别优选使用本发明的挠性基板。作为显示元件、受光元件的制造工序之一例,可举出在形成于基板上的聚酰亚胺树脂膜上,形成对于显示元件、受光元件来说所必需的电路和功能层,使用激光照射等已知方法,将聚酰亚胺树脂膜从基板上剥离。例如作为有机EL元件(其为显示元件之一例),图2示出了有机EL元件之一例(顶部发光方式、白色发光有机EL)。在支承基板7上形成聚酰亚胺树脂膜8,在其上进一步形成作为无机膜的气体阻隔层9,在其上形成TFT电路和有机EL发光层等。TFT电路和有机EL发光层等包含TFT10(其包含无定形硅、低温多晶硅(polysilicon)、氧化物半导体等)、及平坦化层11、第一电极12(其由Al/ITO等形成)、白色有机EL发光层14W(其具有被覆第一电极12的端部的绝缘膜13,并包含空穴注入层、空穴传输层、发光层、电子传输层、电子注入层)、以及第二电极15(其由ITO等形成),并利用气体阻隔层16进行密封。通过激光照射等已知方法将聚酰亚胺树脂膜8从支承基板7剥离,由此可以作为有机EL元件使用。含有本发明的聚酰亚胺树脂膜的有机EL元件可以经过至少以下的工序进行制造。工序(1),将含有聚酰亚胺前体和溶剂的聚酰亚胺前体树脂组合物涂布在支承基板上;工序(2),从涂布后的聚酰亚胺前体树脂组合物中除去溶剂;工序(3),将聚酰亚胺前体进行酰亚胺化得到聚酰亚胺树脂膜;工序(4),在上述聚酰亚胺树脂膜上形成TFT电路和有机EL发光层;工序(5),从上述支承基板剥离上述聚酰亚胺树脂膜。对于上述工序(1)~工序(3)而言,如在<聚酰亚胺树脂膜>中已经详细说明过的那样。上述工序(4)中依次形成TFT10(其包含无定形硅、低温多晶硅、氧化物半导体等)、及平坦化层11、第一电极12(其包含Al/ITO等)、白色或者各色(红色、绿色、蓝色等)的有机EL发光层(其具有被覆第一电极12的端部的绝缘膜13,并包含空穴注入层、空穴传输层、发光层、电子传输层、电子注入层)、第二电极15(其包含ITO等)。此时优选在聚酰亚胺树脂膜8上预先形成作为无机膜的气体阻隔层9,然后形成TFT电路和有机EL发光层,另外,也优选在形成有机EL发光层后,利用气体阻隔层16密封。上述工序(5)与上述情况相同。需要说明的是,光提取方式可以为在TFT基板侧提取光的底部发光方式,也可以为在密封基板侧提取光的顶部发光方式。如上所述,通过使用本发明的聚酰亚胺树脂膜,能够以高精度形成TFT,并且能够减小斜视野时的颜色漂移。进而,即使于高温也可以在本发明的聚酰亚胺树脂膜上形成气体阻隔层,因此能够减少面板的气体透过率。因此,通过使用包含本发明的聚酰亚胺树脂膜的有机EL元件,可以得到黑点等缺陷等少、另外色度不变化、显示品质/显示可靠性高、挠性的有机EL显示器。<有机EL显示器>含有本发明的聚酰亚胺树脂膜的有机EL元件、及/或含有本发明的聚酰亚胺树脂膜的CF可以优选以具备它们的有机EL显示器的形式使用。A.具备有机EL元件(其含有本发明的聚酰亚胺树脂膜)的有机EL显示器如前项<显示元件、受光元件>所记载的那样,通过使用有机EL元件(其使用了本发明的聚酰亚胺树脂膜),能够得到有机EL显示器。例如,因为本发明的聚酰亚胺树脂膜具有高透明性,所以通过在本发明的聚酰亚胺树脂膜上制作气体阻隔膜/TFT电路/有机EL发光层(红色/绿色/蓝色),能够得到图7所示的有机EL显示器(底部发光型)。B.具备CF(其含有本发明的聚酰亚胺树脂膜)的有机EL显示器通过组合CF(其含有本发明的聚酰亚胺树脂膜)和有机EL元件,能够得到全彩色显示的有机EL显示器。特别优选将白色发光有机EL元件(将聚酰亚胺树脂膜用于基材)和本发明的CF组合。需要说明的是,作为用作有机EL元件基材的聚酰亚胺树脂膜,可以为现有的聚酰亚胺树脂膜,也可以为本发明的聚酰亚胺树脂膜。将本发明的CF与白色发光型有机EL元件进行贴合而成的有机EL显示器之一例示于图3。作为其制造工序之一例,可举出以下方法。通过上述的制造方法,在第1支承基板(未图示)上形成本发明的CF20。通过上述方法,另行在第2支承基板(未图示)上形成有机EL元件30(其以聚酰亚胺树脂膜作为基板)。之后,介由粘合层17将CF20和有机EL元件30贴合。之后,分别从支承基板侧对第1、第2支承基板照射激光,由此分别剥离第1、第2支承基板。粘合层没有特别限定,例如,可举出利用光、热使粘合剂、粘接剂、粘结剂固化而成的层。粘合层的树脂没有特别限定,例如,可举出丙烯酸树脂、环氧树脂、聚氨酯树脂、聚酰胺树脂、聚酰亚胺树脂、聚硅氧烷树脂等。形成在第2基板上的聚酰亚胺树脂膜没有特别限定,可以为本发明的聚酰亚胺树脂膜,也可以为已知的聚酰亚胺树脂膜。作为已知的聚酰亚胺,例如,可举出以均苯四甲酸二酐、3,3’,4,4’-联苯四甲酸二酐为酸成分、以对苯二胺、4,4’-二氨基二苯基醚、2,2’-二甲基联苯胺、2,2’-二(三氟甲基)-4,4’-二氨基联苯为二胺成分的全芳香族聚酰亚胺。第1、第2支承基板的剥离中使用的激光的种类没有特别限定,只要能够剥离即可,例如,可举出准分子激光(波长248、308、351nm)、YAG激光(波长1064、532、355nm)、He-Ne激光(633nm)、二氧化碳激光(波长1060nm)等。实施例以下举出实施例等说明本发明,但本发明不限于这些例子。(1)聚酰亚胺树脂膜的制作-1使用TokyoElectronLtd.制涂布显影装置Mark-7,将清漆状态的聚酰亚胺前体树脂组合物旋涂于6英寸的镜面硅晶片(mirrorsiliconwafer),使得在140℃×4分钟的预焙后的膜厚成为15±0.5μm。之后,使用相同的Mark-7的加热板,进行140℃×4分钟的预焙处理(主要从涂布后的聚酰亚胺前体树脂组合物中除去溶剂的工序)。使用惰性气体烘箱(KoyoThermoSystemsCo.,Ltd.制INH-21CD)在氮气流下(氧浓度20ppm以下),以3.5℃/min的速度将预焙膜升温至300℃,保持30分钟,将聚酰亚胺前体进行酰亚胺化,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜。接着,浸渍于氢氟酸中1~4分钟,将聚酰亚胺树脂膜从基板上剥离,进行风干得到聚酰亚胺树脂膜。(2)聚酰亚胺树脂膜的制作-2在切断为1/4的6英寸硅基板上旋涂聚酰亚胺前体,使得140℃×4分钟预焙后的膜厚成为30±1.0μm,除此之外,通过与聚酰亚胺树脂膜的制作-1同样的方法制作聚酰亚胺树脂膜。(3)聚酰亚胺树脂膜(玻璃基板上)的制作-1使用MikasaCo.,Ltd.制的旋涂机MS-A200,在50mm×50mm×0.7mm厚的玻璃基板(旭硝子(株)制AN-100)上旋涂清漆,使得140℃×4分钟的预焙后的膜厚成为15±0.5μm。之后,使用DainipponScreenMfg.Co.,Ltd.制加热板D-SPIN进行140℃×4分钟的预焙处理。使用惰性气体烘箱(KoyoThermoSystemsCo.,Ltd.制INH-21CD)在氮气流下(氧浓度20ppm以下),以3.5℃/min的速度将预焙膜升温至300℃,保持30分钟,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜(玻璃基板上)。(4)聚酰亚胺树脂膜(玻璃基板上)的制作-2使用狭缝涂布机(TorayEngineeringCo.,Ltd.制),在300mm×350mm×0.7mm厚的玻璃基板(旭硝子(株)制AN-100)上旋涂清漆,使得140℃×4分钟的预焙后的膜厚成为15±0.5μm。之后,使用加热板,进行140℃×4分钟的预焙处理。使用惰性气体烘箱(KoyoThermoSystemsCo.,Ltd.制INH-21CD),在氮气流下(氧浓度20ppm以下)经70分钟将预焙膜升温至300℃,保持30分钟,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜(玻璃基板上)。(5)光透过率(T)的测定使用紫外可见光分光光度计((株)岛津制作所制MultiSpec1500),测定400nm处的光透过率。需要说明的是,测定时使用(3)中制作的位于玻璃基板上的聚酰亚胺树脂膜。(6)折射率、双折射的测定使用棱镜耦合器(METRICON公司制、PC2010),测定波长632.8nm的TE折射率(n(TE))及TM折射率(n(TM))。n(TE)、n(TM)分别是相对于聚酰亚胺膜面而言的平行方向、垂直方向的折射率。平均折射率n(AV)由((2×n(TE)2+n(TM)2)/3)^0.5算出,双折射以n(TE)与n(TM)之差即(n(TE)-n(TM))的形式计算。需要说明的是,测定时使用(2)中制作的聚酰亚胺树脂膜。(7)CTE的测定使用热机械分析装置(SIINanoTechnologyInc.制EXSTAR6000TMA/SS6000),在氮气流下进行测定。升温方法按照以下条件进行。第1阶段以升温速度5℃/min升温至200度,除去试样的吸附水;第2阶段以降温速度5℃/min空气冷却至室温。第3阶段以升温速度5℃/min进行本测定,求出50℃~200℃的CTE的平均值。需要说明的是,测定时使用(1)中制作的聚酰亚胺树脂膜。(8)裂缝评价通过(3)中记载的方法制作预焙膜,使用光学显微镜(Nikon制、OPTIPHOT300)通过目视来观察100片,记录产生了裂缝的片数。(9)基板翘曲的测定就翘曲测定而言,载置在MitutoyoCorporation制精密石定盘(1000mm×1000mm)上,针对试验板的4边的各中点及各顶点共计8处,使用量隙规测定自定盘浮起的量(距离)。将所述平均值作为翘曲量。测定于室温进行。(10)黑矩阵的位置精度(BM位置精度)的测定在按照(3)的方法制作的位于玻璃基板上的聚酰亚胺树脂膜上旋涂下述制备例3中制作的黑糊剂,利用加热板于130℃干燥10分钟,形成黑色的树脂涂膜。旋涂正型光致抗蚀剂(Shipley公司制、“SRC-100”),利用加热板于120℃预焙5分钟。接着,介由掩模使用超高压汞灯以100mJ/cm2照射紫外线后,使用2.38%的四甲基氢氧化铵水溶液,同时进行光致抗蚀剂的显影和树脂涂膜的蚀刻,形成图案。进而,利用甲基溶纤剂乙酸酯剥离抗蚀剂后,利用加热板于280℃加热10分钟,由此进行酰亚胺化,形成黑矩阵。测定黑矩阵的厚度,结果为1.4μm。对于所制作的滤色片的黑矩阵偏离理想格((ideallattice))的偏离量,使用SMIC-800(SokkiaTopconCo.,Ltd.制),针对各带玻璃的滤色片基板分别在24点处进行测定。通过计算求出通过测定得到的偏离量的绝对值的平均值,将得到的值作为其水准下的黑矩阵偏离理想格的偏离量。(11)EL元件的色坐标(x,y)的角度依存性测定在按照(4)的方法制作的位于玻璃基板上的聚酰亚胺树脂膜表面,使用ITO靶(TotoLtd.制)进行溅射,由此形成膜厚为160nm的ITO膜。接着,将由此形成的ITO膜在Ar气氛下于200℃加热处理1小时,由此将ITO膜进行退火,接着进行蚀刻处理,在聚酰亚胺基板上形成阳极。使用“SemicoClean56”(商品名,FuruuchiChemicalCorporation制)将得到的基板进行超声波清洗15分钟,然后利用超纯水清洗。将该基板在即将制作元件前进行UV-臭氧处理1小时,设置在真空蒸镀装置内,进行排气直至装置内的真空度为5×10-4Pa以下。通过电阻加热法依次蒸镀空穴传输层、有机发光层、电子传输层,设置红色有机EL发光层。接着,在基板上方的整个面形成包含Mg/ITO的阴极。进而,通过CVD制膜来形成SiON密封膜。将得到的上述基板从蒸镀机中取出,从玻璃基板侧照射准分子激光(波长308nm),由此从玻璃基板剥离有机EL元件。利用6V的恒定电压来驱动所得的有机EL元件,使用亮度取向特性测定装置C9920-11(HamamatsuPhotonicsK.K.制)测定0°、70°方向处的色坐标(x,y)。各方向所测定的(x,y)之差越小,表示斜视野中的颜色漂移越小。以下,记载在实施例中使用的化合物的简称。BPDA:3,3’,4,4’-联苯四甲酸二酐ODPA:3,3’,4,4’-氧双邻苯二甲酸二酐6FDA:4,4’-(六氟异亚丙基)二邻苯二甲酸酐BSAA:2,2-双(4-(3,4-二羧基苯氧基)苯基)丙烷二酐PMDA-HS:1R,2S,4S,5R-环己烷四甲酸二酐BPAF:4,4’-(芴基)二邻苯二甲酸酐BPF-EPA:4,4’-((9H-芴基)双(4,1-亚苯基氧基羰基))二邻苯二甲酸二酐BPF-PA:9,9-双(4-(3,4-二羧基苯氧基)苯基)芴的酸酐CHDA:反式-1,4-二氨基环己烷TFMB:2,2’-双(三氟甲基)联苯胺FDA:9,9-双(4-氨基苯基)芴FDH:9,9-双(3-(3-氨基苯酰胺)-4-羟基苯基)芴BPF-AN:9,9-双(4-(4-氨基苯氧基)苯基)芴NMP:N-甲基-2-吡咯烷酮。制备例1:聚酰亚胺前体组合物(清漆)的制备在干燥氮气流下,在2000mL四颈瓶中加入129.0416g(0.438mol)BPDA、47.4290g(0.438mol)PDA、1000gNMP,于65℃加热搅拌。6小时后,进行冷却得到聚酰胺酸树脂溶液(树脂的浓度15wt%)。制备例2:聚酰亚胺前体组合物(清漆)的制备将60.07g(0.30mol)DAE、70.29g(0.65mol)PDA及12.43g(0.05mol)SiDA与850g的GBL及850g的NMP一同加入,添加309.43g(0.9975mol)ODPA,于80℃反应3小时。添加马来酸酐1.96g(0.02mol),进而于80℃反应1小时,得到聚酰胺酸树脂溶液(树脂的浓度20wt%)。制备例3:用于形成黑矩阵的黑色遮光剂组合物的制作在制备例2的聚酰亚胺前体组合物(清漆)250g中混合50g炭黑(MA100;三菱化学(株)制)及200gNMP,使用Dyno-MillKDL-A,使用直径0.3mm的氧化锆珠,以3200rpm分散处理3小时,得到遮光剂分散液1。在50g该遮光剂分散液1中添加49.9g的NMP及0.1g的表面活性剂(LC951;楠本化学(株)制),得到非感光性的遮光剂组合物。制备例4:用于形成红色着色像素的感光性红色着色剂组合物的制作作为着色剂,混合50gPR177(Chromofine(注册商标)Red6125EC;大日精化制)及50gPR254(Irgaphor(注册商标)RedBK-CF;CibaSpecialtyChemicals制)。在该着色剂中混合100g高分子分散剂(BYK2000;树脂浓度40wt%;BYKJapanKK制)、67g碱可溶性树脂(Cyclomer(注册商标)ACA250;树脂浓度45wt%;DaicelChemicalIndustries,Ltd.制)、83g丙二醇单甲基醚及650g丙二醇单甲基醚乙酸酯,制作浆液。利用管线将加入了浆液的烧杯连接于循环式珠磨分散机(Dyno-MillKDL-A;WillyA.Bachofen制),作为介质使用直径0.3mm的氧化锆珠,以3200rpm分散处理4小时,得到着色剂分散液。在45.7g该着色剂分散液中添加7.8gCyclomerACA250、3.3g光聚合性单体(Kayarad(注册商标)DPHA;日本化药制)、0.2g光聚合引发剂(Irgacure(注册商标)907;CibaSpecialtyChemicals制)、0.1g光聚合引发剂(Kayacure(注册商标)DETX-S;日本化药制)、0.03g表面活性剂(BYK333;BYKJapanKK.制)及42.9g丙二醇单甲基醚乙酸酯,得到着色剂组合物。着色剂组合物的全部固态成分中的着色剂的浓度为31wt%,各着色剂的重量混合比为PR177:PR254=50:50。制备例5:用于形成绿色着色像素的感光性绿色着色剂组合物的制作作为着色剂,混合65gPG7(Hostaperm(注册商标)GreenGNX;ClariantJapanK.K.制)及35gPY150(E4GNGT;LanxessK.K.制)。在该着色剂中混合100gBYK2000、67gCyclomerACA250、83g丙二醇单甲基醚及650g丙二醇单甲基醚乙酸酯,使用Dyno-MillKDL-A,使用直径0.3mm的氧化锆珠,以3200rpm进行6小时分散处理,得到着色剂分散液。在51.7g该着色剂分散液中添加6.3gCyclomerACA250、2.9gKayaradDPHA、0.2gIrgacure907、0.1gKayacureDETX-S、0.03gBYK333及38.8g丙二醇单甲基醚乙酸酯,得到着色剂组合物。着色剂组合物的全部固态成分中的着色剂的浓度为35wt%,着色剂为PG7∶PY150=65∶35(重量比)。制备例6:用于形成蓝色着色像素的感光性蓝色着色剂组合物的制作作为着色剂,使用100gPB15∶6(Lionol(注册商标)Blue7602;ToyoInkCo.,Ltd.制),在该着色剂中混合100gBYK2000、67gCyclomerACA250、83g丙二醇单甲基醚及650g丙二醇单甲基醚乙酸酯,制作浆液。使用分散机Dyno-MillKDL-A,使用直径0.3mm的氧化锆珠,以3200rpm对浆液进行3小时分散处理,得到着色剂分散液。在41.3g该着色剂分散液中,添加8.9gCyclomerACA250、3.5gKayaradDPHA、0.2gIrgacure907、0.1gKayacureDETX-S、0.03gBYK333及46g丙二醇单甲基醚乙酸酯,得到着色剂组合物。着色剂组合物的全部固态成分中的着色剂的浓度为28wt%,着色剂仅为PB15∶6。制备例7:用于形成红色着色像素的非感光性红色着色剂组合物的制作混合3.6g(80wt%)PigmentRedPR254、0.9g(20wt%)PigmentRedPR177、和22.5g聚合物分散剂(PD)及63gNMP,制作浆液。使用分散机Dyno-MillKDL-A,使用直径0.3mm的氧化锆珠,以3200rpm对浆液进行3小时分散处理,得到着色剂分散液。在45.6g该着色剂分散液中,添加制备例2中制作的聚酰亚胺前体清漆18.2g、作为密合改良剂的3-氨基丙基三乙氧基硅烷0.1g、作为表面活性剂的丙烯酸系表面活性剂0.03g、及适量NMP进行混合,得到颜料/树脂比率为25/75(wt/wt)、固态成分浓度为6%且含有94wt%的NMP作为溶剂的红色糊剂(PR-1)。制备例8:用于形成绿色着色像素的非感光性绿色着色剂组合物的制作混合2.7g(60wt%)PigmentGreenPG36、1.8g(40wt%)PigmentYellowPY150、和22.5g聚合物分散剂(PD)及63gNMP,制作浆液。使用分散机Dyno-MillKDL-A,使用直径0.3mm的氧化锆珠,以3200rpm对浆液进行3小时分散处理,得到着色剂分散液。在45.6g该着色剂分散液中添加制备例2中制作的聚酰亚胺前体清漆18.2g、作为密合改良剂的3-氨基丙基三乙氧基硅烷0.1g、作为表面活性剂的丙烯酸系表面活性剂0.03g、及适量NMP进行混合,得到颜料/树脂比率为25/75(wt/wt)、固态成分浓度为6%且含有94wt%的NMP作为溶剂的绿色糊剂(PG-1)。制备例9:用于形成蓝色着色像素的非感光性蓝色着色剂组合物的制作混合4.5gPigmentBluePB15∶6和22.5g聚合物分散剂(PD)及63gN-甲基吡咯烷酮,制作浆液。使用分散机Dyno-MillKDL-A,使用直径0.3mm的氧化锆珠,以3200rpm对浆液进行3小时分散处理,得到着色剂分散液。在45.6g该着色剂分散液中添加制备例2中制作的聚酰亚胺前体清漆18.2g、作为密合改良剂的3-氨基丙基三乙氧基硅烷0.1g、作为表面活性剂的丙烯酸系表面活性剂0.03g、及适量NMP进行混合,得到颜料/树脂比率为25/75(wt/wt)、固态成分浓度为6wt%且含有94wt%的NMP作为溶剂的蓝色糊剂(PB-1)。制备例10:用于形成透明保护膜的树脂组合物的制作在65.05g偏苯三酸中添加280g的GBL及74.95g的γ-氨基丙基三乙氧基硅烷,于120℃加热2小时。在得到的溶液20g中添加7g双苯氧基乙醇芴二缩水甘油醚及15g二乙二醇二甲基醚,得到树脂组合物。制备例11:聚硅氧烷溶液的合成在500ml的三颈瓶中装入甲基三甲氧基硅烷81.72g(0.60mol)、苯基三甲氧基硅烷59.49g(0.30mol)、(2-(3,4-环氧基环己基)乙基三甲氧基硅烷24.64g(0.10mol)、二丙酮醇163.1g,于室温一边搅拌一边经10分钟添加在水55.8g中溶解有磷酸0.54g(相对于装入的单体而言为0.3wt%)的磷酸水溶液。之后,将烧瓶浸在40℃油浴中搅拌30分钟,然后经30分钟将油浴升温至115℃。升温开始1小时后溶液内温达到100℃,自此开始加热搅拌1.5小时(内温为100~110℃)得到聚硅氧烷溶液。需要说明的是,加热搅拌中以0.051(升)/min流过氮。反应中馏出作为副产物的甲醇、水,共计131g。得到的聚硅氧烷溶液的固态成分浓度为43wt%、聚硅氧烷的重均分子量为4200。需要说明的是,聚硅氧烷中的苯基取代硅烷的含有比以Si原子摩尔比计为30mol%。制备例12:感光性正型透明抗蚀剂的制作在黄色灯下混合上述合成中得到的聚硅氧烷溶液15.43g、醌二叠氮化合物0.59g、作为溶剂的二丙酮醇3.73g、丙二醇单甲基醚乙酸酯9.84g,搅拌制成均匀溶液,然后用0.45μm的过滤器进行过滤制备感光性正型透明抗蚀剂。(实施例1)在干燥氮气流下,在200mL四颈瓶中加入3.1495g(27.51mmol)CHDA、1.0651g(3.06mmol)FDA、7.1951g(24.45mmol)BPDA、2.7159g(6.11mmol)6FDA、80gNMP于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时单体的摩尔比为BPDA/6FDA/CHDA/FDA=80/20/90/10。由得到的聚酰亚胺前体通过上述(1)~(4)记载的各方法制作聚酰亚胺树脂膜,通过(5)~(11)记载的方法,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例2)在干燥氮气流下,在200mL四颈瓶中加入3.4068g(29.83mmol)CHDA、0.5471g(1.57mmol)FDA、8.0387g(27.32mmol)BPDA、2.1250g(4.08mmol)BSAA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/BSAA/CHDA/FDA=87/13/95/5。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例3)在干燥氮气流下,在200mL四颈瓶中加入3.4463g(30.18mmol)CHDA、0.9145g(2.62mmol)FDA、7.7216g(26.24mmol)BPDA、2.0353g(6.56mmol)ODPA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/ODPA/CHDA/FDA=80/20/92/8。与实施例1同样地制作固化膜,针对透过率、双折射及线膨胀系数进行测定。结果示于表1。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例4)在干燥氮气流下,在200mL四颈瓶中加入3.7100g(32.49mmol)CHDA、0.7226g(2.62mmol)FDA、8.1354g(27.65mmol)BPDA、1.5496g(6.91mmol)PMDA-HS、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/PMDA-HS/CHDA/FDA=80/20/94/6。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例5)在干燥氮气流下,在200mL四颈瓶中加入3.5082g(30.72mmol)CHDA、0.5634g(1.62mmol)FDA、8.5635g(29.11mmol)BPDA、1.4825g(3.23mmol)BPAF、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/BPAF/CHDA/FDA=90/10/95/5。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例6)在干燥氮气流下,在200mL四颈瓶中加入3.3253g(29.12mmol)CHDA、0.5340g(1.53mmol)FDA、8.1168g(27.59mmol)BPDA、2.1415g(3.07mmol)BPF-EPA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/BPF-EPA/CHDA/FDA=90/10/95/5。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例7)在干燥氮气流下,在200mL四颈瓶中加入3.3662g(29.48mmol)CHDA、0.5406g(1.55mmol)FDA、8.2168g(27.93mmol)BPDA、1.9940g(3.10mmol)BPF-PA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/BPF-PA/CHDA/FDA=90/10/95/5。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例8)在干燥氮气流下,在200mL四颈瓶中加入3.3209g(29.08mmol)CHDA、0.5333g(1.53mmol)FDA、6.7551g(22.96mmol)BPDA、3.5083g(7.65mmol)BPAF、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/BPAF/CHDA/FDA=75/25/95/5。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例9)在干燥氮气流下,在200mL四颈瓶中加入3.1226g(27.35mmol)CHDA、1.1439g(3.04mmol)MFDA、7.1516g(24.31mmol)BPDA、2.6995g(6.08mmol)6FDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/6FDA/CHDA/MFDA=80/20/90/10。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例10)在干燥氮气流下,在200mL四颈瓶中加入3.0978g(27.13mmol)CHDA、1.4594g(2.36mmol)FHA、6.9406g(23.59mmol)BPDA、2.6199g(5.90mmol)6FDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/6FDA/CHDA/FHA=80/20/92/8。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例11)在干燥氮气流下,在200mL四颈瓶中加入3.0211g(26.46mmol)CHDA、1.5657g(2.94mmol)BPF-AN、6.9191g(23.52mmol)BPDA、2.6118g(5.88mmol)6FDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/6FDA/CHDA/BPF-AN=80/20/90/10。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例12)在干燥氮气流下,在200mL四颈瓶中加入3.3598g(29.42mmol)CHDA、1.1391g(3.27mmol)FDA、9.6187g(32.69mmol)BPDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/CHDA/FDA=100/90/10。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例13)在干燥氮气流下,在200mL四颈瓶中加入3.3381g(29.23mmol)CHDA、1.2229g(3.25mmol)MFDA、9.5566g(32.48mmol)BPDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/CHDA/MFDA=100/90/10。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例14)在干燥氮气流下,在200mL四颈瓶中加入3.1619g(27.69mmol)CHDA、1.9035g(3.08mmol)FHA、9.0522g(30.77mmol)BPDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/CHDA/FHA=100/92/8。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(实施例15)在干燥氮气流下,在200mL四颈瓶中加入3.2224g(28.22mmol)CHDA、1.6701g(3.14mmol)BPF-AN、9.2252g(31.35mmol)BPDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/CHDA/BPF-AN=100/90/10。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、裂缝评价、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(比较例1)在干燥氮气流下,在200mL四颈瓶中加入3.9472g(34.57mmol)CHDA、10.1704g(34.57mmol)BPDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(比较例2)在干燥氮气流下,在200mL四颈瓶中加入5.9138g(18.47mmol)TFMB、8.2039g(18.47mmol)6FDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(比较例3)在干燥氮气流下,在100mL四颈瓶中加入3.9014g(34.17mmol)CHDA、7.0366g(23.92mmol)BPDA、3.1796(10.25)ODPA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/ODPA/CHDA=70/30/100。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(比较例4)在干燥氮气流下,在200mL四颈瓶中加入1.8178g(15.92mmol)CHDA、3.6978(10.61mmol)FDA、6.2448g(21.22mmol)BPDA、2.3572g(5.31mmol)6FDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。此时,单体的摩尔比为BPDA/6FDA/CHDA/FDA=80/20/60/40。与实施例1同样地制作聚酰亚胺树脂膜,针对光透过率、双折射、CTE、基板翘曲、BM位置精度、EL元件的色坐标的角度依存性进行测定。结果示于表1。(比较例5)在干燥氮气流下,在200mL四颈瓶中加入7.6544g(21.97mmol)FDA、6.4633g(21.97mmol)BPDA、80gNMP,于65℃加热搅拌。6小时后,冷却制成聚酰亚胺前体组合物(清漆)。与实施例1同样地制作聚酰亚胺树脂膜,结果整个膜中产生裂缝。(实施例16)滤色片的制作(图4)[1]聚酰亚胺树脂膜的制作在300mm×400mm×0.7mm厚的玻璃基板(AN100(旭硝子(株)))(未图示)上旋涂在实施例1中合成的清漆,以使140℃×20分钟的预焙后的厚度成为15±0.5μm。之后,使用加热板,进行140℃×4分钟的预焙处理。使用惰性气体烘箱在氮气流下(氧浓度20ppm以下)以3.5℃/min将预焙处理后的涂膜升温至300℃,保持30分钟,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜2(玻璃基板上)。[2]黑矩阵的制作在通过上述方法制作的位于玻璃基板上的聚酰亚胺树脂膜上旋涂在制备例3中制作的黑糊剂,利用加热板于130℃干燥10分钟,形成黑色的树脂涂膜。旋涂正型光致抗蚀剂(Shipley公司制、“SRC-100’),利用加热板于120℃预焙5分钟。接着,介由掩模使用超高压汞灯,以100mJ/cm2照射紫外线后,使用2.38%的四甲基氢氧化铵水溶液,同时进行光致抗蚀剂的显影和树脂涂膜的蚀刻,形成图案。进而,用甲基溶纤剂乙酸酯剥离抗蚀剂后,利用加热板于280℃加热10分钟,由此使其酰亚胺化,形成黑矩阵4。测定黑矩阵的厚度,结果为1.4μm。[3]着色像素的制作在黑矩阵经过了图案加工的位于玻璃基板上的聚酰亚胺树脂膜上,调整旋转器的旋转数,将在制备例4中制作的感光性红色抗蚀剂涂布在聚酰亚胺膜上,以使热处理后的黑矩阵开口部的厚度成为2.0μm。接着,利用加热板于100℃预焙10分钟,由此得到红色着色像素。接着,使用CanonInc.制紫外线曝光机“PLA-5011”,针对黑矩阵开口部和黑矩阵上的一部分区域,介由以岛状透过光的铬制光掩模,按照100mJ/cm2(365nm处的紫外线强度)进行曝光。曝光后,浸渍在由0.2wt%四甲基氢氧化铵水溶液形成的显影液中进行显影,接着利用纯水进行清洗。之后,利用230℃的烘箱加热处理30分钟,制作红色像素5R。同样地操作,制作由在制备例5中制作的感光性绿色抗蚀剂形成的绿色像素5G、由在制备例6中制作的感光性蓝色抗蚀剂形成的蓝色像素5B。接着,调整旋转器的旋转数,涂布在制备例10中制作的树脂组合物,以使热处理后的着色像素部的厚度成为2.5μm。之后,利用230℃的烘箱加热处理30分钟,制作保护层6。[4]滤色片从玻璃基板上的剥离在通过上述方法在玻璃基板上制作的滤色片的周边刻入切口,在水中浸渍12小时,由此将滤色片从玻璃基板上剥离。需要说明的是,使用光学显微镜确认像素图案形状,结果在剥离前后图案形状未变化。另外,关于得到的滤色片的外观,与玻璃基板滤色片相比,毫不逊色。(实施例17)滤色片的制作着色像素的制作中,使用制备例7~9所记载的非感光性着色剂组合物,通过下述记载方法制作着色像素,除此之外,与实施例16同样地制作滤色片。在黑矩阵4经过了图案加工的位于玻璃基板上的聚酰亚胺树脂膜2上,调整旋转器的旋转数,将制备例7中制作的非感光性着色剂组合物涂布在聚酰亚胺膜上,以使热处理后的黑矩阵开口部的厚度成为2.0μm。接着,利用加热板于130℃干燥10分钟,形成红色的树脂涂膜。在红色的树脂涂膜上旋涂正型光致抗蚀剂(Shipley公司制、“SRC-100”),利用加热板于120℃预焙5分钟。接着,使用超高压汞灯,按照100mJ/cm2照射紫外线进行掩模曝光后,使用2.38%的四甲基氢氧化铵水溶液,同时进行光致抗蚀剂的显影和树脂涂膜的蚀刻,形成图案。利用甲基溶纤剂乙酸酯剥离抗蚀剂,利用加热板于280℃加热10分钟,由此使其酰亚胺化,形成红色像素5R。同样地操作,制作由在制备例8中制作的非感光性绿色着色剂组合物形成的绿色像素5G、由在制备例9中制作的非感光性蓝色着色剂组合物形成的蓝色像素5B。接着,调整旋转器的旋转数,涂布在制备例10中制作的树脂组合物,以使热处理后的着色像素部的厚度成为2.5μm。之后,在230℃的烘箱中加热处理30分钟,制作保护层6。使用光学显微镜,确认得到的滤色片的像素图案形状,结果在剥离前后图案形状未变化。另外,关于得到的滤色片的外观,与玻璃基板滤色片相比,毫不逊色。(实施例18)滤色片的制作(图5)使用由氧化硅形成的靶,在氩气氛下进行溅射,在聚酰亚胺树脂膜2上形成厚度300nm的由氧化硅膜形成的气体阻隔层3,在该层合膜上形成黑矩阵4、着色像素5R、5G、5B、保护层6,除此之外,与实施例17同样地制作滤色片。在压力2×10-1Pa、基板温度300℃、交流电源13.56MHz的条件下通过溅射形成气体阻隔层。使用光学显微镜确认像素图案形状,结果在剥离前后图案形状未变化。另外,关于得到的滤色片的外观,与玻璃基板滤色片相比毫不逊色。(实施例19)滤色片的制作(图6)使用氧化锌、二氧化硅与氧化铝的比率为62/35/3(mol)的混合烧结靶,在含有10vol%氧的氩气氛下进行溅射,在聚酰亚胺树脂膜2上形成厚度200nm的由氧化硅/氧化锌/氧化铝膜形成的气体阻隔层(下层)3’,之后,在维持真空的状态下,使用由氧化硅形成的靶,在氩气氛下进行溅射,得到厚度100nm的由氧化硅膜形成的气体阻隔层(上层)3”。在该层合膜上形成黑矩阵4、着色像素5R、5G、5B、保护层6,除此之外,与实施例17同样地制作滤色片。在压力3×10-1Pa、基板温度300度、直流电源3kW的条件下通过溅射形成气体阻隔膜(下层)。在压力2×10-1Pa、基板温度300℃、交流电源13.56MHz的条件下通过溅射形成气体阻隔层(上层)。使用光学显微镜确认像素图案形状,结果在剥离前后图案形状未变化。另外,关于得到的滤色片的外观,与玻璃基板滤色片相比毫不逊色。(实施例20)有源矩阵型有机EL元件的制作(图7)[1]聚酰亚胺树脂膜的制作在300mm×400mm×0.7mm厚的玻璃基板(AN100(旭硝子(株)))(未图示)上旋涂在实施例1中制备的清漆,以使140℃×10分钟的预焙后的厚度成为15±0.5μm。之后,使用送风干燥器进行140℃×10分钟的预焙处理。使用惰性气体烘箱在氮气流下(氧浓度20ppm以下)以3.5℃/min将预焙处理后的涂膜升温至300℃,保持30分钟,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜2(玻璃基板上)。[2]TFT基板的制作在通过上述方法制作的聚酰亚胺树脂膜(玻璃基板上)上,使用等离子体CVD法形成由SiO形成的气体阻隔层9。之后,形成底栅型的TFT10,以覆盖该TFT的状态形成由Si3N4形成的绝缘膜(未图示)。接着,在该绝缘膜上形成接触孔,然后在绝缘膜上形成介由该接触孔与TFT连接的配线(高度1.0μm,未图示)。该配线用于将TFT之间连接、或者用于将在后续工序中形成的有机EL元件与TFT连接。进而,为了将由配线的形成而导致的凹凸进行平坦化,在埋入由配线引起的凹凸的状态下向绝缘膜上形成平坦化层11。平坦化层的形成如下进行:将感光性聚酰亚胺清漆旋涂在基板上,在加热板上进行预焙(120℃×3分钟)后,介由所期望的图案的掩模进行曝光、显影,在空气流下于230℃加热处理60分钟。涂布清漆时的涂布性良好,曝光、显影、加热处理后得到的平坦化层中未确认到折皱、裂缝的产生。进而,配线的平均阶差为500nm,制作的平坦化层中形成5μm见方的接触孔,厚度为约2μm。[3]底部发光型有机EL元件的制作在得到的平坦化层11上形成以下各部位,制作底部发光型有机EL元件。首先,在平坦化层上形成由ITO形成的第一电极12,使其介由接触孔与配线(未图示)连接。之后,涂布抗蚀剂并进行预焙,介由所期望的图案的掩模进行曝光、显影。将该抗蚀剂图案作为掩模,通过使用了ITO蚀刻剂的湿蚀刻进行第一电极的图案加工。之后,使用抗蚀剂剥离液(单乙醇胺与二乙二醇单丁基醚的混合液)剥离该抗蚀剂图案。将剥离后的基板进行水洗,于200℃加热脱水30分钟得到带平坦化层的电极基板。关于平坦化层的厚度尺寸变化,相对于剥离液处理前、在加热脱水后小于1%。由此得到的第一电极相当于有机EL元件的阳极。接着,形成覆盖第一电极的端部的形状的绝缘层13。绝缘层使用相同的感光性聚酰亚胺清漆。通过设置该绝缘层,可以防止第一电极与后续工序中形成的第二电极15之间的短路。进而,在真空蒸镀装置内介由所期望的图案掩模,依次蒸镀空穴传输层、有机发光层、电子传输层,设置红色有机EL发光层14R、绿色有机EL发光层14G、蓝色有机EL发光层14B。接着,在基板上方的整个面形成包含Al/Mg(Al:反射电极)的第二电极15。进而通过CVD成膜形成SiON密封膜16。从蒸镀机中取出所得到的上述基板,从玻璃基板侧照射准分子激光(波长308nm),由此从玻璃基板上剥离有机EL元件。介由驱动电路向得到的有源矩阵型有机EL元件施加电压,结果显示良好的发光。另外,得到的有机EL元件与使用玻璃基板制作的有机EL元件相比毫不逊色。(实施例21)有机EL显示器(有源矩阵型)的制作(图8)[1]聚酰亚胺树脂膜的制作在300mm×400mm×0.7mm厚的玻璃基板(AN100(旭硝子(株)))上旋涂实施例1中制备的清漆,以使140℃×10分钟的预焙后的厚度成为15±0.5μm。之后,使用送风干燥器进行140℃×10分钟的预焙处理。使用惰性气体烘箱在氮气流下(氧浓度20ppm以下)以3.5℃/min将预焙处理后的涂膜升温至300℃,保持30分钟,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜2(玻璃基板上)。[2]TFT基板的制作在通过上述方法制作的聚酰亚胺树脂膜(玻璃基板上)上,采用等离子体CVD法形成由SiO形成的气体阻隔层9。之后,形成底栅型TFT10,以覆盖该TFT10的状态形成由Si3N4形成的绝缘膜(未图示)。接着,在该绝缘膜上形成接触孔,然后在绝缘膜上形成介由该接触孔与TFT连接的配线(高度1.0μm,未图示)。该配线用于将TFT之间连接、或者用于将在后续工序中形成的有机EL元件与TFT连接。进而,为了将由配线的形成而导致的凹凸进行平坦化,在埋入由配线引起的凹凸的状态下向绝缘膜上形成平坦化层11。平坦化层如下形成:将感光性聚酰亚胺清漆旋涂在基板上,在加热板上进行预焙(120℃×3分钟)后,介由所期望的图案的掩模进行曝光、显影,在空气流下于230℃加热处理60分钟,由此形成平坦化层。涂布清漆时的涂布性良好,曝光、显影、加热处理后得到的平坦化层中未确认到折皱、裂缝的产生。进而,配线的平均阶差为500nm,制作的平坦化层中形成5μm见方的接触孔,厚度为约2μm。[3]滤色片层的制作在上述平坦化层11上,调整旋转器的旋转数,涂布在制备例7中制作的非感光性红色着色剂组合物PR-1,以使热处理后的厚度成为1.9μm,利用120℃的加热板加热10分钟,由此得到红色着色像素。使用狭缝涂布机涂布正型光致抗蚀剂(RohmandHaasElectronicMaterialsK.K.制、“LC-100A’),以使预焙后的厚度成为1.0μm,利用100℃的加热板,干燥5分钟,进行预焙。使用CanonInc.制紫外线曝光机PLA-501F,介由光掩模以100mJ/cm2(365nm的紫外线强度)进行掩模曝光。接着,使用2.0%的四甲基氢氧化铵水溶液,同时进行光致抗蚀剂的显影和树脂涂膜的蚀刻,形成图案。接着,利用甲基溶纤剂乙酸酯剥离抗蚀剂。接着,在270℃的烘箱热中处理30分钟,由此进行固化,制作厚度1.9μm的红色像素5R。同样地操作,使用在制备例8中制作的非感光性绿色着色剂组合物PG-1形成绿色像素5G,使用在制备例9中制作的非感光性蓝色着色剂组合物PB-1形成蓝色像素5B。[3]保护层的制作使用旋涂机(MikasaCo.,Ltd.制1H-360S)以任意的旋转数将制备例12中制作的感光性正型透明抗蚀剂旋涂在形成有滤色片层的基板上,然后使用加热板(DainipponScreenMfg.Co.,Ltd.制SCW-636)于90℃预焙2分钟,制作厚度3μm的膜。使用平行光光刻机(以下,简称为PLA)(CanonInc.制PLA-501F),使用超高压汞灯,介由用于测定灵敏度的灰度掩模对制作的膜进行图案曝光,然后使用自动显影装置(TakizawaSangyoCo.,Ltd.制AD-2000)喷淋2.38重量%四甲基氢氧化铵水溶液即ELM-D(商品名,三菱气体化学(株)制)60秒进行显影,接着,用水冲洗30秒。之后,作为漂白曝光,使用PLA(CanonInc.制PLA-501F),使用超高压汞灯以3000J/m2(换算为波长365nm曝光量)对膜的整个面进行曝光。之后,使用加热板,于110℃软烘焙(softbaking)2分钟,接着使用烘箱(TabaiEspecCorp.制IHPS-222)在空气中于230℃固化1小时,制作保护层(未图示)。[4]底部发光型有机EL元件的制作在得到的保护层上形成以下各部位,制作底部发光型有机EL元件。首先,在保护层上形成由ITO形成的第一电极12,使其介由接触孔与配线(未图示)连接。之后,涂布抗蚀剂并进行预焙,介由所期望的图案的掩模进行曝光、显影。将该抗蚀剂图案作为掩模,通过使用了ITO蚀刻剂的湿蚀刻进行第一电极12的图案加工。之后,使用抗蚀剂剥离液(单乙醇胺与二乙二醇单丁基醚的混合液)剥离该抗蚀剂图案。将剥离后的基板进行水洗,于200℃加热脱水30分钟得到带滤色片层的电极基板。关于滤色片层的厚度尺寸变化,相对于剥离液处理前而言,在加热脱水后小于1%。由此得到的第一电极相当于有机EL元件的阳极。接着,形成覆盖第一电极12的端部的形状的绝缘层13。绝缘层13中使用相同的感光性聚酰亚胺清漆。通过设置该绝缘层13,可以防止第一电极与后续工序中形成的第二电极15之间的短路。进而,在真空蒸镀装置内介由所期望的图案掩模,依次蒸镀空穴传输层、有机发光层、电子传输层,设置白色有机EL发光层14W。接着,在基板上方的整个面形成由Al/Mg(Al:反射电极)形成的第二电极15。进而通过CVD成膜形成SiON密封膜16。从蒸镀机中取出所得到的上述基板,从玻璃基板侧照射准分子激光(波长308nm),由此从玻璃基板上剥离有机EL元件。介由驱动电路向得到的有源矩阵型有机EL元件施加电压,结果显示良好的发光。另外,得到的有机EL元件与使用玻璃基板制作的有机EL元件相比毫不逊色。(实施例22)有机EL显示器(有源矩阵型)的制作(图9)[1]聚酰亚胺树脂膜的制作在300mm×400mm×0.7mm厚的玻璃基板(AN100(旭硝子(株)))上旋涂实施例1中制备的清漆,以使140℃×10分钟的预焙后的厚度成为15±0.5μm。之后,使用送风干燥器进行140℃×10分钟的预焙处理。使用惰性气体烘箱在氮气流下(氧浓度20ppm以下)以3.5℃/min将预焙处理后的涂膜升温至300℃,保持30分钟,以5℃/min的速度冷却至50℃,制作聚酰亚胺树脂膜2(玻璃基板上)。[2]TFT基板的制作在通过上述方法制作的聚酰亚胺树脂膜2(玻璃基板上)上,采用等离子体CVD法形成由SiO形成的气体阻隔层9。之后,形成底栅型TFT10,以覆盖该TFT的状态形成由Si3N4形成的绝缘膜(未图示)。接着,在该绝缘膜上形成接触孔,然后在绝缘膜上形成介由该接触孔与TFT连接的配线(高度1.0μm,未图示)。该配线用于将TFT之间连接、或者用于将在后续工序中形成的有机EL元件与TFT连接。进而,为了将由配线的形成而导致的凹凸进行平坦化,在埋入由配线引起的凹凸的状态下向绝缘膜上形成平坦化层11。平坦化层如下形成:将感光性聚酰亚胺清漆旋涂在基板上,在加热板上进行预焙(120℃×3分钟)后,介由所期望的图案的掩模进行曝光、显影,在空气流下于230℃加热处理60分钟,由此形成平坦化层。涂布清漆时的涂布性良好,曝光、显影、加热处理后得到的平坦化层中未确认到折皱、裂缝的产生。进而,配线的平均阶差为500nm,制作的平坦化层中形成5μm见方的接触孔,厚度为约2μm。[3]顶部发光型有机EL元件的制作在通过上述方法得到的TFT的平坦化层11上形成以下各部位,制作顶部发光型有机EL元件。首先,在平坦化层11上形成由Al/ITO(Al:反射电极)形成的第一电极12,使其介由接触孔与配线连接。之后,涂布抗蚀剂并进行预焙,介由所期望的图案的掩模进行曝光、显影。将该抗蚀剂图案作为掩模,通过使用了ITO蚀刻剂的湿蚀刻进行第一电极的图案加工。之后,使用抗蚀剂剥离液(单乙醇胺与二乙二醇单丁基醚的混合液)剥离该抗蚀剂图案。将剥离后的基板进行水洗,于200℃加热脱水30分钟得到带平坦化层的电极基板。关于平坦化层的厚度变化,相对于剥离液处理前而言,在加热脱水后小于1%。由此得到的第一电极12相当于有机EL元件的阳极。接着,形成覆盖第一电极12端部的形状的绝缘层13。绝缘层使用相同的感光性聚酰亚胺清漆。通过设置该绝缘层,能够防止第一电极12与后续工序中形成的第二电极15之间的短路。进而,在真空蒸镀装置内介由所期望的图案掩模,依次蒸镀空穴传输层、有机发光层、电子传输层,设置红色有机EL发光层14R、绿色有机EL发光层14G、蓝色有机EL发光层14B。接着,在基板上方的整个面形成由Mg/ITO形成的第二电极15。进而通过CVD成膜形成SiON密封膜16。从蒸镀机中取出所得到的上述基板,从玻璃基板侧照射准分子激光(波长308nm),由此从玻璃基板上剥离有机EL元件。介由驱动电路向得到的有源矩阵型有机EL元件施加电压,结果显示良好的发光。另外,得到的有机EL元件与使用玻璃基板制作的有机EL元件相比毫不逊色。(实施例23)有机EL显示器(有源矩阵型)的制作(图3)[1]带玻璃基板的滤色片的制作通过实施例18记载的方法,在玻璃基板上制作滤色片。[2]带玻璃基板的TFT的制作使用在制备例1中制作的聚酰亚胺前体清漆,除此之外,通过实施例20中记载的方法,依次制作聚酰亚胺树脂膜8、气体阻隔层9、TFT10、平坦化层11。[3]顶部发光型有机EL元件的制作在通过上述方法得到的TFT的平坦化层11上形成以下各部位,制作顶部发光型有机EL元件。首先,在平坦化层11上形成由Al/ITO(Al:反射电极)形成的第一电极12,使其介由接触孔与配线连接。之后,涂布抗蚀剂进行预焙,介由所期望的图案的掩模进行曝光、显影。将该抗蚀剂图案作为掩模,通过使用了ITO蚀刻剂的湿蚀刻进行第一电极12的图案加工。之后,使用抗蚀剂剥离液(单乙醇胺与二乙二醇单丁基醚的混合液)剥离该抗蚀剂图案。将剥离后的基板进行水洗,于200℃加热脱水30分钟得到带平坦化层的电极基板。关于平坦化层的厚度变化,相对于剥离液处理前而言,在加热脱水后小于1%。由此得到的第一电极12相当于有机EL元件的阳极。接着,形成覆盖第一电极12端部的形状的绝缘层13。绝缘层使用相同的感光性聚酰亚胺清漆。通过设置该绝缘层,可以防止第一电极与后续工序中形成的第二电极15之间的短路。进而,在真空蒸镀装置内介由所期望的图案掩模,依次蒸镀空穴传输层、有机发光层、电子传输层,设置白色有机EL发光层14W。接着,在基板上方的整个面形成由Mg/ITO形成的第二电极15。进而通过CVD成膜形成SiON密封膜16。[4]有机EL显示器的制作将上述[1]中得到的带玻璃基板的滤色片与[3]中得到的带玻璃基板的顶部发光型有机EL元件介由粘合层17进行贴合。接着,从玻璃基板侧照射准分子激光(波长308nm),由此从玻璃基板上剥离滤色片和有机EL元件,制作有机EL显示器。介由驱动电路在得到的有源矩阵型有机EL显示器上施加电压,结果显示良好的发光。另外,得到的有机EL元件与使用玻璃基板制作的有机EL元件相比毫不逊色。符号说明1支承基板(玻璃基板)2聚酰亚胺树脂膜3气体阻隔层3’气体阻隔层(下层)3”气体阻隔层(上层)4黑矩阵5R红色着色像素5G绿色着色像素5B蓝色着色像素6保护层7玻璃基板8聚酰亚胺树脂膜9气体阻隔层10TFT11平坦化层12第一电极13绝缘层14W白色有机EL发光层14R红色有机EL发光层14G绿色有机EL发光层14B蓝色有机EL发光层15第二电极16密封膜17粘合层20CF30有机EL元件[表1]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种聚酰亚胺复合材料及其制备方法与制造工艺
- 树脂前体及含有其的树脂组合物、聚酰亚胺树脂膜、树脂薄膜及其制造方法与制造工艺
- 使用了2‑苯基‑4,4’‑二氨基二苯基醚类的清漆、成形性优异的酰亚胺树脂组合物及具有优异断裂伸长率的固化树脂成形体、以及使用了这些的预浸料、酰亚胺预浸料、及耐热性和机械强度优异的纤维强化材料的制造方法与工艺
- 球状氧化铝粉末以及使用该球状氧化铝粉末的树脂组成物的制造方法与工艺
- 用于高TG聚酰亚胺覆铜板的树脂材料配方的制造方法与工艺
- 一种BPADA型13BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种HQDA型14BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种BPADA型14BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种PMDA型双酚A四胺支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种ODPA型13BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺