用于在作物中确定供体插入的快速靶向分析的制作方法
本申请基于35U.S.C.§119(e)要求获得于2013年9月4日提交的美国临时专利申请No.61/873,719和于2013年11月4日提交的美国临时专利申请No.61/899,569的利益,其全部内容通过提述并入本申请中。
对电子提交的序列列表的援引
序列表的正式拷贝作为ASCII格式的序列列表通过EFS-Web电子递交,文件名为“226007_ST25.txt”,生成日期为2013年11月04日,大小为68.6千字节,并且与本申请一并提交。本ASCII格式文档中包含的序列列表是专利申请的一部分,其全部内容通过提述并入本申请中。
发明领域
本公开一般地涉及分子生物学和生物化学领域。本公开涉及用于分析被整合的供体多核苷酸的基因组插入位点的方法。该方法可用于高通量分析被整合的供体多核苷酸,并能够用于最小化检测到的假阳性结果。而且,本方法使用基于细胞的靶向和分析,不需要产生稳定靶向的植物。
发明背景
植物的靶向基因组修饰已经成为应用研究和基础研究的一个长期未能达到且难以达到的目标。将基因和基因堆叠靶向到植物基因组中的特定位置将会改善转基因事件的质量,降低产生转基因事件的相关成本,并提供制造转基因植物产品的新方法,例如顺序性基因堆叠。总的来说,将转基因靶向特定基因组位点很可能是商业上有利的。过去几年中,开发方法和组合物,用于通过位点特异性核酸酶(例如锌指核酸酶(ZFN)、大范围核酸酶、转录激活物样效应物核酸酶(TALENS)、以及成簇规则间隔短回文重复/CRISPR-相关核酸酶(CRISPR/Cas)结合工程化crRNA/tracr RNA)靶向和切割基因组DNA,以诱导靶向突变、诱导细胞DNA的靶向删除、以及易化外源供体DNA多核苷酸在预定的基因组座位内的靶向重组,已经取得了显著的进展。参见例如,美国专利公开号20030232410;20050208489;20050026157;20050064474;和20060188987,和国际专利公开号WO 2007/014275,将它们的公开通过提述并入本申请用于所有目的。美国专利公开号20080182332描述了非经典锌指核酸酶(ZFN)用于植物基因组的靶向修饰的用途,美国专利公开号20090205083描述了植物EPSP基因组座位的ZFN介导的靶向修饰。现有的外源DNA靶向插入的方法涉及用含有至少一种转基因的供体DNA多核苷酸与位点特异性核酸酶(例如ZFN)共转化植物组织,其中位点特异性核酸酶设计为结合并切割特定的基因组座位。这导致供体DNA多核苷酸稳定地插入被切割的基因组座位内,结果实现规定基因组座位处的靶向基因添加。
不幸的是,已报告和观察到的靶向基因组修饰的频率表明,靶向植物体内的基因座位是相对低效的。已报告的低效率使得为了鉴定含有靶向基因组座位的特定事件需要筛选大量的植物事件。筛选方法还应当适用作为用于快速鉴定含有被靶向的基因组座位的植物事件的高通量方法。此外,因为靶向基因插入与随机基因插入一起出现,所以筛选方法必须被设计为在随机插入的背景中特异性鉴定出被靶向的靶向基因组座位,并将基因组整合与可能产生假阳性结果的外源质粒DNA区分开来。而且,测定法应当灵敏到足以检测发生在单个细胞内的事件,其中该细胞含有在数千个非靶向细胞中才有一个的靶向事件。大多数报道的植物事件分析依靠单一的分析方法来确认靶向,这可能导致对靶向频率的估计不准确并且输出结果的可信度低。需要开发能够检测位点特异性染色体整合并将这些事件与外源质粒DNA区分开的改进的分子测定方法,特别是高通量分析。最后,目前用于评估靶向基因组修饰的方法是基于产生稳定的植物,在时间和成本上是密集的。因此,需要这样一种分析方法,其允许快速靶向评估大量的基因组座位并筛选大量的位点特异性核酸酶,以鉴定和确认多核苷酸供体序列在靶基因组座位内的插入。
前述的相关技术及其相关限制的实施例旨在举例说明而非是排他性的。本领域技术人员通过阅读本专利说明书对相关技术的其它限制将显而易见。
发明概要
在一个实施方案中,本公开涉及一种用于检测多核苷酸供体序列在基因组靶位点内的位点特异性整合的测定,其中:通过第一轮PCR,使用设计为结合基因组DNA靶位点的第一外-PCR引物和设计为结合被整合的多核苷酸供体序列的第一内-PCR引物来扩增基因组DNA,产生第一扩增子;和通过第二轮PCR,使用特异针对位于第一扩增子内部的序列的引物来扩增上述第一扩增引物,产生第二扩增子;并检测该第二扩增子的存在,其中第二扩增子的产生指示位点特异性整合事件的存在。
在该实施方案的一个方面中,基因组靶位点包括内源的或工程化的基因组靶位点。在该实施方案的另一个方面中,以低于第一外-PCR引物的浓度提供第一内-PCR引物。在一个实施方案中,实施第一轮PCR所使用的第一外-PCR引物对第一内-PCR引物的相对浓度为大约4:1,3:1或2:1。在另一个实施方案中,第一内-PCR引物的浓度为0.05–0.09μM,且第一外-PCR引物的浓度为至少0.1μM。
在该实施方案的下一个方面,上述第二轮PCR包括设计为结合所述第一扩增子的基因组DNA靶位点的第二外-PCR引物,和设计为结合所述第一扩增子的被整合的多核苷酸供体序列的第二内-PCR引物。在一个实施方案中,实施第二轮PCR使用的第二外-PCR引物对第二内-PCR引物的相对浓度为大约4:1、3:1或2:1。在进一步的实施方案中,第二内-PCR引物的浓度为0.05–0.1μM,且第二外-PCR引物的浓度为至少0.2μM。
在该实施方案的进一步的方面中,包含位点特异性整合在基因组靶位点内的多核苷酸供体序列的基因组DNA是植物基因组DNA。作为一个实施方案,植物基因组DNA从单子叶植物分离得到。作为另一个实施方案,植物基因组DNA从双子叶植物分离得到。
在该实施方案的另一个方面中,用位点特异性核酸酶切割基因组DNA靶位点导致多核苷酸供体序列在该基因组靶位点内的位点特异性整合。作为一个实施方案,位点特异性核酸酶选自下组:锌指核酸酶、CRISPR核酸酶、TALEN核酸酶、或大范围核酸酶(meganuclease)。在下一个实施方案中,多核苷酸供体序列在基因组靶位点内的位点特异性整合是通过非同源末端连接(Non Homologous End Joining)机制发生的。
在该实施方案的一个方面中,检测步骤是对第二扩增子进行琼脂糖凝胶电泳或者对第二扩增子进行测序反应。
在该实施方案的另外一个方面中,本公开涉及用于检测多核苷酸供体序列在被转染的植物细胞基因组靶位点内的位点特异性整合的方法,包括:通过第一轮PCR扩增基因组DNA产生第一扩增子,其中所述PCR使用设计为结合该基因组靶位点的第一外-PCR引物和设计为结合该多核苷酸供体序列的第一内-PCR引物来实施,进一步其中所述第一内-PCR引物以低于该第一外-PCR引物的浓度提供;通过第二轮PCR使用特异针对位于所述第一扩增子内部的序列的引物来扩增该第一扩增引物,产生第二扩增子;以及检测该第二扩增子的存在,其中第二扩增子的产生指示位点特异性整合事件的存在。在其它实施方案中,植物细胞是原生质体植物细胞。在一个实施方案中,位点特异性整合的检测对靶向植物细胞与非靶向植物细胞(targeted and non-targeted plant cells)的混合群体进行,其中非靶向植物细胞在基因组靶位点内不含有多核苷酸供体序列。
除了上面描述的示例性方面和实施方案之外,进一步的方面和实施方案将通过研究下面的说明书变得显而易见。
附图简述
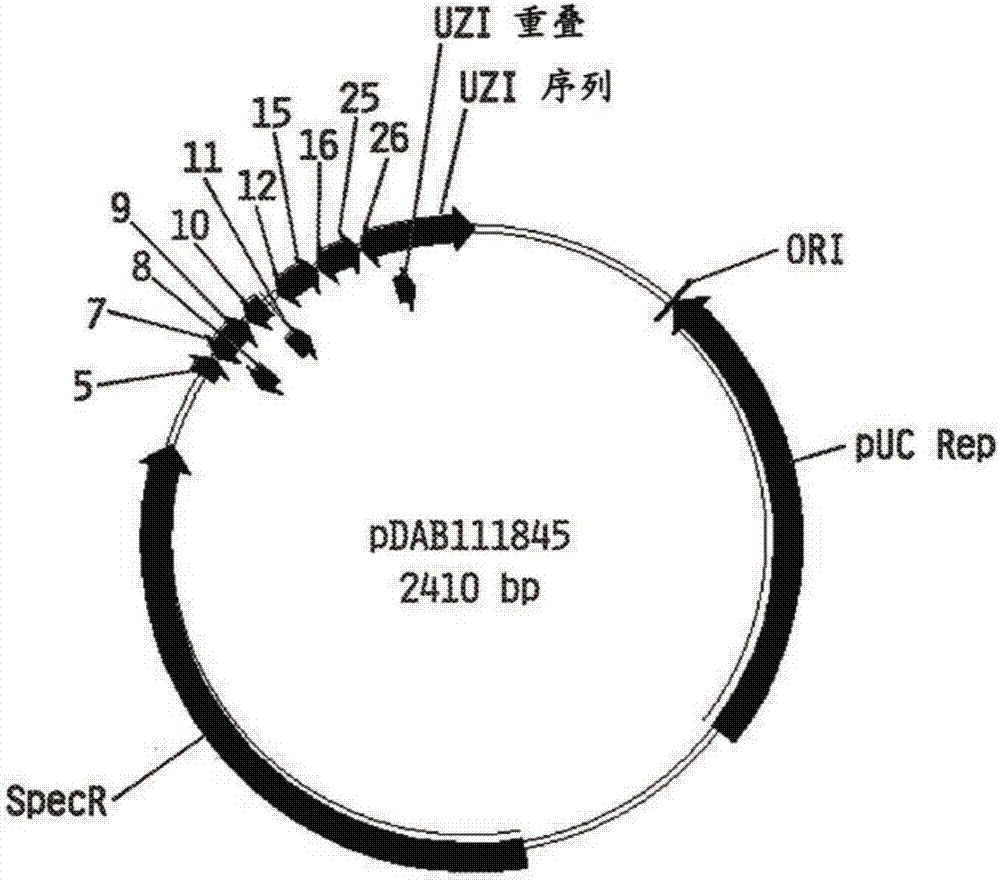
图1图示pDAB111845的质粒图。
图2图示pDAB111846的质粒图。
图3图示pDAB117415的质粒图。
图4图示pDAB117416的质粒图。
图5图示pDAB117417的质粒图。
图6图示pDAB117419的质粒图。
图7图示pDAB117434的质粒图。
图8图示pDAB117418的质粒图。
图9图示pDAB117420的质粒图。
图10图示pDAB117421的质粒图。
图11图示用于通过NHEJ进行整合的通用供体多核苷酸序列的示意图。
图12图示用于通过HDR进行整合的通用供体多核苷酸序列的示意图。标签“HA”指示同源臂;标签“ZFN BS”指示ZFN结合位点(用于单体)。
图13图示用于靶向和验证通用供体多核苷酸系统整合在玉米选定基因组座位内的靶向验证的构建体。A)ZFN设计空间,标出了ZFN对的位置。B)ZFN表达构建体的配置。标记“NLS”指示核定位信号,标记“ZFP”指示锌指蛋白。C)用于NHEJ介导靶向玉米选定基因组座位的通用供体多核苷酸。Z1-Z6代表特异性针对玉米选定基因组座位靶标的ZFN结合位点。ZFN位点的数目可以在3-6之间变化。垂直箭头显示独特的限制位点,水平箭头代表潜在的PCR引物位点。该通用供体多核苷酸系统是所有用于在玉米选定基因组座位内整合的供体所共有的一个短(110bp)序列。
图14图示pDAB8393的质粒图。
图15A和15B图示玉米选定基因组座位靶点处的ZFN切割活性。切割活性用每100万个高品质读段中在ZFN切割位点处具有插入缺失(Indel)的序列的数目来表示。图15A用柱状图形式显示了数据。图15B用表格显示数据。
图16图示使用基于NHEJ的快速靶向分析方法对玉米选定基因组座位靶标的验证。
图17图示通过随机整合转化到玉米内的质粒构建体,它们包括用于侧翼序列分析和转基因表达研究的事件。
图18图示在原生质体快速靶向分析中在ELP处介由NHEJ的供体插入。插入可以沿着正向或反向发生。
图19图示ELP1中ZFN切割位点的破坏。破坏用qPCR信号的降低表示,以靶对参考的比值为单位。对于ZFN1和ZFN3分别观察到平均22%和15%的信号降低。
图20图示内-外扩增的PCR产物的序列。对每个内-外PCR的4个克隆进行测序,结果证明了完整的靶供体接点和被加工的末端接点。所列出的序列的对应关系为:SEQ I NO:248对应于“预测”,SEQ ID NO:249对应于A9-1,SEQ ID NO:250对应于A9-2,SEQ ID NO:251对应于A9-5,SEQ ID NO:252对应于A9-6,SEQ ID NO:253对应于G8-1,SEQ ID NO:254对应于G8-2,SEQ ID NO:255对应于G8-5,SEQ ID NO:256对应于G8-6,SEQ ID NO:257对应于G9-1,SEQ ID NO:258对应于G9-2,SEQ ID NO:259对应于G9-6,SEQ ID NO:260对应于H9-1,SEQ ID NO:261对应于H9-2,SEQ ID NO:262对应于H9-5,SEQ ID NO:263对应于H9-6。
图21图示在原生质体快速靶向分析中在E32处介由NHEJ的供体插入。插入可以沿着正向或反向发生。
图22是显示设计用于供体多核苷酸的引物与锌指结合序列之关系的示意图。
图23是pDAB7221的质粒图。
图24是用于座位破坏分析的探针/引物示意图。标明了FAD2 2.3和2.6基因的F2ZFN结合位点和用于破坏测试的引物。
图25提供了在FAD2 2.3基因座内使用F2,ZFN2锌指核酸酶进行供体序列之NHEJ靶向而产生的内-外PCR产物的序列。参考序列(图的顶部)代表供体载体沿着反方向靶向插入的配置。填充通过FokI消化产生的DNA的单链末端以产生参考序列。显示了桑格法测序序列(Sanger sequence)。F2、ZFN2ZFN结合序列用下划线指示。在右侧列出了与指定序列具有相似序列的质粒克隆。
详细说明书
I.概览
现在公开了用于快速筛选、鉴定和表征特异性核酸酶靶标植物事件的新方法。这些方法能够用于通过第一和第二扩增反应分析基因组靶位点内供体多核苷酸的整合。第一和第二扩增反应是一种“内-外”PCR扩增反应,用于筛选基因组座位内被靶定的供体DNA多核苷酸的3’和/或5’接点序列。含有3’和/或5’接点序列的扩增产物的存在表明供体DNA多核苷酸存在于靶标基因组座位内。
所公开的筛选测定描述了用于鉴定和获得靶转基因插入事件的高品质、高通量的过程。通过应用该筛选测定,可以对大量植物事件进行分析和筛选,以选择在靶基因组座位内插入了供体DNA多核苷酸的特定事件,并将这些事件与假阳性结果区分开来。而且,本公开的方法可以用作高通量测定,能够快速而高效地鉴定出样品的能进一步通过其它分子确认方法分析的子集。本公开的主题包括植物和植物细胞,其包含利用这种新筛选方法选出的核酸酶靶植物事件。而且,本方法可容易地用于任何植物物种的分析。
II.术语
除非另外指出,否则这里所用的全部技术和科学术语具有与本公开所涉及领域的普通技术人员所公知的相同的含义。当存在矛盾时,以本申请(包括定义)作准。除非上下文另外要求,否则单数形式的术语应包括多数的形式,多数形式的术语应包括单数的形式。本文提到的全部公开、专利和其它参考文献的全部内容通过提述并入本公开中用作所有目的,就如同每一个单独的公开或专利申请被具体且单独地指明通过提述并入一样,除非明示仅将专利或专利申请公布的特定部分通过提述并入本文。
在了进一步阐明本公开,提供了下面的术语、缩写和定义。
如本文所使用的,术语“包括”、“包含”、“含有”、“具有”或其任何其它变体均意图是非排他性的或者是开放式的。例如,包含一系列元件的组合物、混合物、过程、方法、物品、或装置并不比仅限制于那些元件,而是可以包含其它没有表达列出的或者这些组合物、混合物、过程、方法、物品或装置所固有的其它元件。而且,除非有明确地相反陈述,否则“或”是指包含性的“或”,而不是排他性的“或”。例如,条件A或B通过如下的任何一项而满足:A为真(或存在)且B为假(或不存在),A为假(或不存在)且B为真(或存在),A和B都为真(或存在)。
如本文所使用的,术语“发明”或“本发明”是一个非限制性的术语,并不意图指某一发明的任何一个实施方案,而是包括本申请中公开的全部可能的实施方案。
如本文所使用的,术语“植物”包括整个植物和任何后代、植物的细胞、组织、或部分。术语“植物部分”包括植物的任何部分,包括,例如但不限于:种子(包括成熟种子、不成熟的种子、和没有种皮的未成熟胚);植物插条(plant cutting);植物细胞;植物细胞培养物;植物器官(例如,花粉、胚、花、果实、芽、叶、根、茎,和相关外植体)。植物组织或植物器官可以是种子、愈伤组织、或者任何其他被组织成结构或功能单元的植物细胞群体。植物细胞或组织培养物能够再生出具有该细胞或组织所来源的植物的生理学和形态学特征的植物,并能够再生出与该植物具有基本上相同基因型的植物。与此相反,一些植物细胞不能够再生产生植物。植物细胞或组织培养物中的可再生细胞可以是胚、原生质体、分生细胞、愈伤组织、花粉、叶、花药、根、根尖、丝、花、果仁、穗、穗轴、壳、或茎。
植物部分包括可收获的部分和可用于繁殖后代植物的部分。可用于繁殖的植物部分包括,例如但不限于:种子;果实;插条;苗;块茎;和砧木。植物的可收获部分可以是植物的任何有用部分,包括,例如但不限于:花;花粉;苗;块茎;叶;茎;果实;种子;和根。
植物细胞是植物的结构和生理单元。如本文所使用的,植物细胞包括原生质体和具有部分细胞壁的原生质体。植物细胞可以处于分离的单个细胞或细胞聚集体的形式(例如,松散愈伤组织和培养的细胞),并且可以是更高级组织单元(例如,植物组织、植物器官、和植物)的一部分。因此,植物细胞可以是原生质体、产生配子的细胞,或者能够再生成完整植物的细胞或细胞的集合。因此,在本文的实施方案中,包含多个植物细胞并能够再生成为整株植物的种子被认为是一种“植物部分”。
如本文所使用的,术语“原生质体”是指细胞壁被完全或部分地除去、其脂双层膜裸露的植物细胞。典型地,原生质体是没有细胞壁的分离植物细胞,其具有再生成细胞培养物或整株植物的潜力。
如本文所使用的,“内源序列”定义了位于生物体或生物体基因组中天然位置处的多核苷酸、基因或多肽的自然形式。
如本文所使用的,术语“分离的”意思是从其自然环境中移出。
如本文所使用的,术语“纯化的”是指分子或化合物以基本上游离于天然或自然环境下通常与该分子或化合物相关联的污染物的形式分开,并且意味着由于与原始组合物的其他组分分开而导致纯度增加。术语“纯化的核酸”在本文中用于描述这样的核酸序列:其与其他化合物(包括但不限于多肽、脂质和碳水化合物)分开。
如本文所使用的,术语“多核苷酸”、“核酸”和“核酸分子”可互换使用,并且可以包括单个核酸;多个核酸;核酸片段、其变异体或衍生物;和核酸构建体(例如,信使RNA(mRNA)和质粒DNA(pDNA))。多核苷酸或核酸可以含有全长cDNA序列或其片段的核苷酸序列,包括非翻译的5’和/或3’序列和编码序列。多核苷酸或核酸可以包括任何多聚核糖核苷酸或多聚脱氧核糖核苷酸,其可以包括未经修饰的核糖核苷酸或脱氧核糖核苷酸或经过修饰的核糖核苷酸或脱氧核糖核苷酸。例如,多核苷酸或核酸可以包括单链和双链DNA;单链区和双链区混合的DNA;单链和双链RNA;和单链区和双链区混合的RNA。包含DNA和RNA的杂交分子可以是单链、双链、或单链和双链区的混合物。前述术语还包括多核苷酸或核酸的化学、酶学和代谢修饰形式。
应当理解,具体的DNA还指其互补物,互补物的序列可以根据脱氧核糖核苷酸碱基配对规则来确定。
如本文所使用的,术语“基因”是指编码功能产物(RNA或多肽/蛋白质)的核酸。基因可以包括位于功能产物编码序列前面(5’非编码序列)和/或后面(3’非编码序列)的调节序列。
如本文所使用的,术语“编码序列”是指编码特定氨基酸序列的核酸序列。“调节序列”是指位于编码序列上游(例如5’非编码序列)、内部或下游(例如3’非编码序列),影响相关编码序列的转录、RNA加工或稳定性、或翻译的核苷酸序列。调节序列包括,例如但不仅限于:启动子;翻译前导序列;内含子;多聚腺苷酸化识别序列;RNA加工位点;效应子结合位点;和茎环结构。
如本文所使用的,术语“多肽”包括单个多肽、多个多肽,及其片段。这个术语是指由单体(氨基酸)通过酰胺键(也称为肽键)线性连接的分子。术语“多肽”是指任何包含两个或多个氨基酸的任何链,并且不指产物的特定长度或尺寸。因此,肽、二肽、三肽、寡肽、蛋白质、氨基酸链,和任何其它用于指示包含两个或多个氨基酸的链的术语,均包含在“多肽”的定义内,并且前述的各术语在本文中可以和“多肽”互换地使用。多肽可以从天然生物来源分离或者通过重组技术生产,但是特定的多肽并不一定是从特定的核酸翻译的。多肽可以任何适当的方式产生,包括例如但不限于通过化学合成。
与之相反,术语“异源的”是指通常不会出现其在参考(宿主)生物体中的位置处的多核苷酸、基因或多肽。例如,异源核酸可以是通常出现在参考生物体的其他基因组位置处的核酸。另举一例,异源核酸可以是通常不会出现在参考生物体中的核酸。可以通过将异源多核苷酸、基因或多肽引入到宿主生物体中而产生包含异源多核苷酸、基因或多肽的宿主生物体。在具体实例中,异源多核苷酸包括以与相应的天然多核苷酸不同的形式被重新引入到来源生物体中的天然编码序列或其部分。在具体实例中,异源基因包括以与相应的天然基因不同的形式被重新引入到来源生物体中的天然编码序列或其部分。例如,异源基因可以包括这样的天然编码序列,其是被重新引入到天然宿主体内的嵌合基因的一部分,所述嵌合基因包含非天然的调节区域。在具体实例中,异源多肽是以不同于相应的天然多肽的形式被重新引入到来源生物体内的天然多肽。
异源基因或多肽可以是这样的基因或多肽,其包含功能性多肽或编码功能性多肽的核酸序列,所述功能性多肽或编码功能性多肽的核酸序列与另一个基因或多肽融合生成嵌合或融合多肽,或者编码它们的基因。具体实施方案的基因和蛋白质包括具体例示的全长序列和部分、节段、片段(包括连续片段和与全长分子相比有内部和/或末端缺失)、变体、突变体、嵌合体、和这些序列的融合体。
如本所使用的,术语“修饰”可以指本文公开的多核苷酸的改变,导致由该多核苷酸编码的多肽的活性减小、基本上消失或消失,以及本文公开的多肽的改变,导致该多肽的活性减小、基本上消失或消失。或者,术语“修饰”可以指本文公开的多核苷酸的改变,导致由该多核苷酸编码的多肽的活性增加或提高,以及本文公开的多肽的改变,导致该多肽的活性增加或提高。这些改变可以通过本领域众所周知的方法实现,包括但不仅限于,删除、突变(例如自发突变、随机突变、由增变基因(mutator gene)导致的突变,或转座子诱变),取代、插入、下调、改变细胞定位、改变多核苷酸和多肽的状态(例如,甲基化、磷酸化或泛素化),除去辅助因子、引入反义RNA/DNA、引入中断RNA/DNA、化学修饰、共价修饰、用紫外线或X射线辐射、同源重组、有丝分裂重组、启动子置换方法,和/或它们的组合。
如本文所使用的,术语“衍生”是指对本公开中提出的序列的修饰。这些修饰的举例包括取代、插入和/或删除一个或多个与本文公开的编码序列的核酸序列相关的碱基,其会导致本文公开的编码序列在作物物种中的功能被保留、略微改变或增加。这些衍生可以被本领域的技术人员容易地确定,例如使用用于预测和优化序列结构的计算机建模技术。术语“衍生物”因此还包括具有与本文公开的编码序列具有基本上相同的序列的核酸序列,从而使它们能够具有用于产生本公开实施方案的被公开的功能性。
术语“启动子”是指能够控制核酸编码序列或功能性RNA表达的DNA序列。在实例中,受控制的编码序列位于启动子序列的3'。启动子可以完全来自天然基因,启动子可以包括来自在自然界中发现的不同启动子的不同元件,或者启动子甚至可以包括合理设计的DNA片段。本领域的技术人员会理解,不同的启动子可以在不同的组织或细胞类型中,或者在不同的发育阶段,或者响应不同的环境或生理条件,而指导基因的表达。上述所有启动子的实例都是已知的,并且在现有技术中被用于控制异源核酸的表达。可以在大部分时间在大多数细胞类型中指导基因表达的启动子通常称为“组成型启动子”。此外,虽然本领域有人试图(在许多情况下未能)划分调节序列的确切边界,但是已经发现,不同长度的DNA片段可能具有相同的启动子活性。具体核酸的启动子活性可以使用本领域人员熟悉的技术加以测定。
术语“可操作连接”是指单个核酸上的多个核酸序列的关联,其中一个核酸序列的功能受到另一个核酸序列的影响。例如,当启动子能够影响编码序列表达时(例如,编码序列在启动子的转录控制之下),启动子与该编码序列是可操作连接的。编码序列可以沿着有义或反义方向与调节序列可操作连接。
如本文所使用的,术语“表达”可以指来自DNA的有义(mRNA)或反义RNA的转录和稳定积累。表达也可以指mRNA翻译成多肽。如本文所使用的,术语“过表达”是指高于相同基因或相关基因的内源表达的表达。因此,如果其表达高于可比较的内源基因,则异源基因“过表达”。
如本文所使用的,术语“转化”或“转染”是指核酸或其片段被转移并整合到宿主生物体中,导致在遗传上稳定的继承。含有转化核酸的宿主生物体被称为“转基因的”、“重组的”或“被转化的”的生物体。已知的转化方法包括,例如:根癌土壤杆菌或发根土壤杆菌介导的转化;磷酸钙转化;凝聚胺转化;原生质体融合;电穿孔;超声方法(例如,超声穿孔(sonoporation));脂质体转化;显微注射;用裸DNA转化;用质粒载体转化;用病毒载体转化;基因枪转化(微粒轰击);碳化硅晶须介导转化;气溶胶发射(aerosol beaming);和PEG介导的转化。
如本文所使用的,术语“引入/导入”(在将核酸引入到细胞内的上下文中)包括细胞转化,以及令含有该核酸的植物与第二植物杂交,从而使第二植物含有该核酸,这可以使用常规植物育种技术来实施。这些育种技术是本领域已知的。关于植物育种技术的讨论参见Poehlman(1995)Breeding Field Crops,第4版,AVI Publication Co.,Westport CT。
回交方法可用于将核酸引入到植物中。这一技术用于将性状引入到植物中已有数十年之久。描述回交(和其他植物育种方法)的一个实例可见,例如,Poelman(1995),同上;和Jensen(1988)Plant Breeding Methodology,Wiley,New York,NY。在示范性回交方案中,感兴趣的原始植物(“轮回亲本”)与携带被引入的核酸的第二植物(“非轮回亲本”)杂交。随后令该杂交得到的子代再次与轮回亲本杂交,并重复该过程直到获得的被转换的植物,其中除了来自非轮回亲本的核酸之外,轮回亲本的基本上所有期望的形态学和生理学特征都在被转换的植物中也得到了恢复。
“结合”是指大分子之间(例如,蛋白质和核酸之间)的序列特异性的、非共价的相互作用。结合相互作用并不要求其所有组分都是序列特异性的(例如,与DNA骨架中的磷酸残基接触),只要该相互作用整体上是序列特异性的即可。这样的相互作用一般被10-6M-1或更低的解离常数(Kd)所表征。“亲和力”是指结合的强度:越高的结合亲和力与越低的Kd相关。
“结合蛋白”是能够与另一个分子非共价结合的蛋白质。结合蛋白能够与,例如,DNA分子(DNA结合蛋白)、RNA分子(RNA结合蛋白)和/或蛋白质(蛋白结合蛋白)结合。在蛋白结合蛋白实例中,它能够与自身结合(形成同源二聚体、同源三聚体等)和/或能够与一个或多个不同的蛋白结合。结合蛋白能够具有超过一种类型的结合活性。例如,锌指蛋白具有DNA结合、RNA结合和蛋白结合活性。
“重组”是指两个多核苷酸之间遗传信息的交换过程,包括但不仅限于:通过非同源末端连接(NHEJ)和同源重组的供体捕获。为本公开的目的,“同源重组(HR)”是指这种交换的特化形式,它在例如细胞中双链断裂修复过程中通过同源介导的修复机制发生。这个过程需要核苷酸序列同源性,使用“供体”分子来模板修复“靶”分子(即,经历了双链断裂的分子),并且有不同称谓如“非交换基因转换(non-crossover gene conversion)”或“短束基因转换(short tract gene conversion)”,这是因为它导致遗传信息从供体转移到靶标。不希望受限于任何特定的理论,这种转移可能涉及在被断裂的靶标与供体之间形成的异源双链DNA的错配校正,和/或“合成依赖性链退火”,其中供体用来重新合成遗传信息并成为靶标的一部分,和/或相关处理。这种特化的HR通常导致靶分子序列发生改变,使得供体多核苷酸的一部分或全部被并入到靶多核苷酸中。对于HR指导的整合,供体分子含有至少一个与基因组同源的区域(“同源臂”),长度至少为50-100个碱基对。参见例如美国专利公开No.20110281361。
在本公开的方法中,一个或多个靶定核酸酶,如本文所描述的,会在预定位点的靶序列(例如细胞染色质)中创造双链断裂,而与断裂区核苷酸序列具有同源性的“供体”多核苷酸会被引入到细胞中。已经显示,双链断裂的存在可以促进供体序列的整合。供体序列可以被物理整合,或者,供体多核苷酸可以被用作模板通过同源重组修复断裂,结果导致与供体中相同的核苷酸序列的全部或一部分被引入到细胞染色质中。由此,细胞染色质中的第一序列可以被改变,并且在某些实施方案中,可以被转换成存在于供体多核苷酸序列内的序列。因此,使用术语“取代”或“替代”可以理解为表示一种核苷酸序列被另一种核苷酸序列替换(即,在信息意义上的序列替换),而不一定要求某个多核苷酸被另一个多核苷酸物理地或化学地替换。
“切割”是指DNA分子共价骨架的断裂。切割可以由多种方法引发,包括但不限于磷酸二酯键的酶学或化学水解。单链切割和双链切割都是可能的,并且双链切割可能作为两个不同的单链切割事件的结果而发生。DNA切割可以导致产生平末端或粘末端(staggered ends)。在某些实施方案中,靶向的双链DNA切割使用融合多肽。
如本文所使用的,术语“质粒”和“载体”是指一种染色体外元件,其可携带一个或多个不是细胞核心代谢之一部分的基因。质粒和载体通常是环形双链DNA分子。然而,质粒和载体可以是线性或环状核酸,作为单链或双链DNA或RNA,并可以携带来自几乎任何来源的DNA,其中多个核苷酸序列已经被连接或重组成一种独特的构造,其能够将启动子片段和编码DNA序列以及任何合适的3’非翻译序列引入到细胞内。在实例中,质粒和载体可以包括自主复制序列,用于在细节宿主中繁殖。
多肽和“蛋白质”在本文可互换使用,包括由通过肽键连接的两个或多个氨基酸残基构成的分子链。该术语不指示产物的特定长度。因此,“肽”和“寡肽”包含在多肽的定义内。该术语包括多肽的翻译后修饰,例如,糖基化、乙酰化、磷酸化等。此外,蛋白质片段、类似物、突变或变体蛋白、融合蛋白等也包含在多肽的含义内。该术语还包括这样的分子,其中包含一个或多个氨基酸类似物或非规范或非天然的氨基酸,其可以被合成,或者使用已知的蛋白质工程化技术重组表达。此外,本发明的融合蛋白可以通过众所周知的有机化学技术,如本文中所描述的,加以衍生化。
术语“融合蛋白”表明蛋白质包含来自多于一种亲本蛋白或多肽的多肽组分。典型地,融合蛋白从融合基因表达而得,融合基因中编码来自一种蛋白的多肽序列的核苷酸序列与另一个核苷酸序列对框附合,且二者任选地可被接头隔开,该另一个核苷酸序列编码来自另一不同蛋白的多肽序列。然后融合基因可被重组宿主细胞表达为单个蛋白质。
III.本发明的实施方案
在一个实施方案中,本公开涉及用于检测多核苷酸供体序列位点特异性整合在基因组靶位点内的测定。
在一些实施方案中,对基因组DNA进行测定,以检测多核苷酸供体序列在基因组靶位点内的位点特异性整合。在该实施方案的各方面中,基因组DNA包括:染色体基因组DNA、线粒体基因组DNA、转座元件基因组DNA、来源于病毒整合的基因组DNA、人工染色体基因组DNA(参见PCT/US2002/017451和PCT/US2008/056993,作为非限制性实例包含在本文中),以及其它来源的基因组DNA。
在一些实施方案中,基因组DNA通过聚合酶链式反应(PCR)被扩增。在本实施方案的各方面中,PCR一般是指用于不经过克隆或纯化而增加基因组DNA的混合物中靶序列节段的浓度的方法(美国专利号中No.4,683,195;4,683,202;和4,965,188;通过引用并入本文)。这个用于扩增靶序列的过程包括,向含有期望靶序列的DNA混合物中引入过量的两种寡核苷酸引物,随后在DNA聚合酶的存在下进行一系列精确的热循环。两个引物与它们各自的双链靶序列的链互补。为了实现扩增,混合物被变性,然后引物与靶分子内的互补序列退火。退火之后,用聚合酶延伸引物,从而形成一对新的互补链。变性、引物退火和聚合酶延伸的步骤可以重复许多次(即,变性、退火和延伸构成一个“循环”;可以有多个“循环”)以获得高浓度的期望靶序列的扩增节段。期望靶序列的扩增节段的长度由引物彼此之间的相对位置决定,因此,这一长度是一个可控的参数。由于该过程的重复性,该方法被称为“聚合酶链式反应”(下文称作“PCR”)。因为靶序列的期望扩增节段成为混合物中的主要序列(就浓度而言),所以称它们为“PCR扩增的”。
在其它实施方案中,PCR反应产生扩增子。作为该实施方案的一个方面,扩增子是指通过延伸一对扩增引物中的任何一个或两个产生的扩增反应的产物。如果所用的两个引物均与靶序列杂交,则扩增子可以含有指数扩增的核酸。或者,如果所用的引物中一个不与靶序列杂交,则扩增子可以通过线性扩增而产生。因此,这个术语在本文中被一般性地使用,并且不一定表示存在指数扩增的核酸。
选定的核酸序列,或靶核酸序列的扩增可以通过任何合适的方法实施。一般地参见Kwohet al.,Am.Biotechnol.Lab.8,14-25(1990)。合适的扩增技术的实例包括,但不仅限于,聚合酶链式反应、连接酶链式反应、链置换扩增(一般地参见G.Walker et al.,Proc.Natl.Acad.Sci.USA 89,392-396(1992);G.Walker et al.,Nucleic Acids Res.20,1691-1696(1992))、基于转录的扩增(参见D.Kwoh et al.,Proc.Natl.Acad Sci.USA 86,1173-1177(1989))、自持序列复制(或"3SR")(参见J.Guatelli et al.,Proc.Natl.Acad.Sci.USA 87,1874-1878(1990))、Qβ复制酶系统(参见P.Lizardi et al.,BioTechnology 6,1197-1202(1988))、基于核酸序列的扩增(或"NASBA")(参见R.Lewis,Genetic Engineering News 12(9),1(1992))、修复链反应(或"RCR")(参见R.Lewis,同上),和回旋镖DNA扩增(boomerang DNA amplification)(或"BDA")(参见R.Lewis,同上)。聚合酶链式反应一般是优选的。
在另一个实施方案中,基因组DNA的扩增通过使用引物的PCR反应完成。在该实施方案的一个方面中,引物可以包括第一组引物、第二引物组、第三组引物,以此类推。这样,指称的“第一”、“第二”、“第三”等表示引物组在巢式PCR反应中被使用的顺序。例如,“第一”组引物最先用于第一轮PCR反应来扩增多核苷酸序列。接着,在第二轮PCR反应中使用“第二”组引物组扩增第一轮PCR反应的产物。然后,在第三轮PCR反应中使用“第三”组引物扩增第二轮PCR反应的产物,以此类推。在本实施方案的其他方面中,引物可以是“外(Out)”引物,其设计为结合基因组DNA靶位点,或者“内(In)”引物,其设计为结合整合在生物体基因组内的多核苷酸供体序列。在其他实施方案中,第一组引物可以包括一个内和一个外引物,或者可以设计为包括两个不同的内引物,或两个不同的外引物。在一个实施方案中,术语“引物”指这样的寡核苷酸,其与在合适的扩增缓冲液中要被扩增的DNA模板互补的。在某些实施方案中,引物的长度可以是10Bp-100Bp,10Bp-50Bp或10Bp-25Bp。
在本主题公开的一个实施方案中,内引物的浓度低于外引物。该实施方案的一个方面包含的外引物与内引物的相对浓度为大约10:1,9:1,8:1,7:1,6:1,5:1,4:1,3:1或2:1。在另一个方面中,该实施方案包含的内引物的浓度为0.001,0.005,0.01,0.02,0.03,0.04,0.05,0.06,0.07.0.008,或0.09μM,而外引物的浓度为至少0.1μM。在进一步的方面中,实施方案包含的内引物的浓度为0.01,0.02,0.03,0.04,0.05,0.06,0.07.0.08,0.09,0.1,0.11 0.12,0.13,0.14,0.15,0.16,0.17,0.18或0.19μM,而外引物的浓度为至少0.2μM。
在一些实施方案中,基因组整合位点是植物基因组DNA。在一个方面中,根据本公开转化的植物细胞包括,但不限于,任何高等植物,既包括双子叶植物也包括单子叶植物,特别是可消费的植物,包括作物植物。这些植物可以包括,但不限于,例如:紫花苜蓿,大豆,棉花,油菜(也称为芥花(canola)),亚麻籽,玉米,水稻,臂形草(brachiaria),小麦,红花,高粱,糖用甜菜,向日葵,烟草和草坪草。因此,任何植物物种或植物细胞均可被选用。在实施方案中,本文所用的植物细胞,由其生长或衍生出的植物包括,但不限于,可以从下列植物中获得的细胞:油菜(Brassica napus);印度芥(Brassica juncea);埃塞俄比亚芥(Brassica carinata);芜菁(Brassica rapa);甘蓝(Brassica oleracea);大豆(Glycine max);亚麻籽/亚麻(Linumusitatissimum);玉蜀黍(也称作玉米(corn))(Zea mays);红花(Carthamus tinctorius);向日葵(Helianthus annuus);烟草(Nicotiana tabacum);拟南芥;巴西坚果(Betholettia excelsa);蓖麻子(Ricinus communis);椰子(Cocusnucifera);芫荽(Coriandrum sativum);棉花(Gossypium spp.);花生(Arachis hypogaea);加州希蒙得木(Simmondsia chinensis);油棕榈(Elaeis guineeis);橄榄(Olea eurpaea);水稻(Oryza sativa);南瓜(Cucurbita maxima);大麦(Hordeum vulgare);甘蔗(Saccharum officinarum);水稻(Oryza sativa);小麦(Triticum spp,包括硬粒小麦(Triticum durum)和普通小麦(Triticum aestivum));和浮萍(Lemnaceae SP)。在一些实施方案中,植物物种中的遗传背景可能会有所不同。
关于遗传修饰植物的产生,对植物进行遗传工程化的方法是本领域已知的。例如,已经开发出了许多用于植物转化的方法,包括用于双子叶植物以及单子叶植物的生物和物理转化方案(例如,Goto-Fumiyuki et al.,Nature Biotech 17:282-286(1999);Miki et al.,Methods in Plant Molecular Biology and Biotechnology,Glick,B.R.and Thompson,J.E.Eds.,CRC Press,Inc.,Boca Raton,pp.67-88(1993))。另外,用于植物细胞或组织转化和植物再生的载体和体外培养方法可以在下列文献中获得,例如Gruber et al.,Methods in Plant Molecular Biology and Biotechnology,Glick,B.R.and Thompson,J.E.Eds.,CRC Press,Inc.,Boca Raton,第89-119页(1993)。
有很多种技术可供用于将DNA插入到植物宿主细胞中。那些技术包括使用根癌土壤杆菌或发根土壤杆菌作为转化剂的卸甲T-DNA(disarmed T-DNA)转化,磷酸钙转染,聚凝胺转化,原生质体融合,电穿孔,超声方法(例如,超声穿孔(sonoporation)),脂质体转化,显微注射,裸DNA,质粒载体,病毒载体,基因枪(微粒轰击),碳化硅晶须介导的转化,气溶胶发射(aerosol beaming),或PEG以及其他可能的方法。
例如,可以使用植物细胞原生质体电穿孔和显微注射等技术把DNA构建体直接引入到植物细胞的基因组DNA中,或者可以使用基因枪方法,例如DNA微粒轰击(参见Klein et al.(1987)Nature 327:70-73),将DNA构建体直接引入到植物组织中。其他用于植物细胞转化的方法包括通过碳化硅晶须介导的DNA摄取进行显微注射(Kaeppler et al.(1990)Plant Cell Reporter9:415-418)。或者,DNA构建体可以通过纳米颗粒转化引入到植物细胞中(参见,例如,美国专利申请号No.12/245,685,其全部内容通过引用并入本文)。
另一种已知的植物转化方法是微粒介导的转化(microprojectile-mediated transformation),其中DNA被携带在微粒的表面上。在这种方法中,使用基因枪设备将载体导入到植物组织中,该设备将微粒加速到足以穿透植物细胞壁和细胞膜的速度。Sanford et al.,Part.Sci.Technol.5:27(1987),Sanford,J.C.,Trends Biotech.6:299(1988),Sanford,J.C.,Physiol.Plant 79:206(1990),Klein et al.,Biotechnology 10:268(1992)。
或者,基因转移和转化方法包括,但不限于,通过氯化钙沉淀、聚乙二醇(PEG)或电穿孔介导的裸DNA摄取进行的原生质体转化(参见Paszkowski et al.(1984)EMBO J 3:2717-2722,Potrykus et al.(1985)Molec.Gen.Genet.199:169-177;Fromm et al.(1985)Proc.Nat.Acad.Sci.USA 82:5824-5828;和Shimamoto(1989)Nature 338:274-276)和植物组织电穿孔(D'Halluin et al.(1992)Plant Cell 4:1495-1505)。
一种广泛使用的将表达载体引入到植物中的方法是基于土壤杆菌的自然转化系统。Horsch et al.,Science 227:1229(1985)。根癌土壤杆菌和发根土壤杆菌是植物致病性土壤细菌,已知可用于对植物细胞进行遗传转化。根癌土壤杆菌的Ti质粒和发根土壤杆菌的Ri质粒分别携带用于植物遗传转化的基因。Kado,C.I.,Crit.Rev.Plant.Sci.10:1(1991)。关于土壤杆菌载体系统和土壤杆菌介导的基因转移的描述也可以从例如Gruber et al.,同上,Miki et al.,同上,Moloney et al.,Plant Cell Reports 8:238(1989),和美国专利号4,940,838和5,464,763获得。
如果使用土壤杆菌进行转化,则应当将待插入的DNA克隆到特定的质粒中,即克隆到中间载体中或者克隆到双元载体中。中间载体自己不能在土壤杆菌中复制。中间载体可以通过使用辅助质粒转移到根癌土壤杆菌中(接合)。日本烟草超级双元系统是这种系统的一个实例(综述参见Komari et al.,(2006),收载于:Methods in Molecular Biology(K.Wang,ed.)No.343:Agrobacterium Protocols(第2版,第1卷)Humana Press Inc.,Totowa,NJ,pp.15-41;和Komori et al.,(2007)Plant Physiol.145:1155-1160)。双元载体既可以在大肠杆菌复制也可以在土壤杆菌中自我复制。它们包括一个选择标记基因和一个接头或多接头,它们被右T-DNA边界区和左T-DNA边界区所界定。可以把它们直接转化至土壤杆菌中(Holsters,1978)。用作宿主细胞的土壤杆菌要包含携带vir区域的质粒。Ti或Ri质粒还包含用于T-DNA转移必需的vir区域。该vir区域是将T-DNA转移到植物细胞内必需的。可以包含其他的T-DNA。
当使用双元T-DNA载体(Bevan(1984)Nuc.Acid Res.12:8711-8721)或非双元T-DNA载体程序(Horsch et al.(1985)Science 227:1229-1231)用根癌土壤杆菌宿主感染植物细胞时,该细菌的毒力功能(virulence function)将指导含有构建体及邻近标记物的T-链插入到植物细胞DNA中。一般地,使用土壤杆菌转化系统对双子叶植物工程化(Bevan et al.(1982)Ann.Rev.Genet16:357-384;Rogers et al.(1986)Methods Enzymol.118:627-641)。土壤杆菌转化系统还可用于将DNA转化和转移到单子叶植物和植物细胞。参见美国专利5,591,616;Hernalsteen et al.(1984)EMBO J 3:3039-3041;Hooykass-Van Slogteren et al.(1984)Nature 311:763-764;Grimsley et al.(1987)Nature325:1677-179;Boulton et al.(1989)Plant Mol.Biol.12:31-40;和Gould et al.(1991)Plant Physiol.95:426-434。在将遗传构建体引入到特定植物细胞之后,可以培育植物细胞,并且在出现分化组织例如芽和根时,可以产生成熟的植物。在一些实施方案中,可以产生多个植物。用于再生植物的方法是本领域普通技术人员已知的,并且可以在例如Plant Cell and Tissue Culture,1994,Vasil和Thorpe编辑.Kluwer Academic Publishers,和Plant Cell Culture Protocols(Methods in Molecular Biology 111,1999Hall编辑,Humana Press中找到。本文所述的遗传修饰植物可以在发酵培养基中培养,或者在合适的培养基,例如土壤中种植。在一些实施方案中,用于高等植物的合适生长培养基可以包括任何用于植物的生长培养基,包括但不限于,土壤、砂、任何其它支持根生长的颗粒介质(例如,蛭石、珍珠岩等)或者水培培养基,以及适当的光、水和优化高等植物生长的营养补充剂。
可以培养通过上述任何一种转化技术产生的转化植物细胞,以再生具有被转化的基因型、并因此具有期望的表型的完整植物。这样的再生技术依赖于对组织培养物生长培养基中某些植物激素的操纵,典型地依赖已经与期望的核苷酸序列一起被引入的杀生物剂和/或除草剂标记。从培养的原生质体再生植物在Evans,et al.,“Protoplasts Isolation and Culture”,收载于Handbook of Plant Cell Culture,第124-176页,Macmillian Publishing Company,New York,1983;和Binding,Regeneration of Plants,Plant Protoplasts,第21-73页,CRC Press,Boca Raton,1985中有描述。也可以从植物愈伤组织、外植体、器官、花粉、胚或其部分获得再生。这样的再生技术在Klee et al.(1987)Ann.Rev.of Plant Phys.38:467-486中有一般地描述。
在其它实施方案中,被转化的植物细胞不能再生产生植物。这样的细胞称作被瞬时转化的。可以产生瞬时转化的细胞用于测定特定转基因的表达和/或功能。瞬时转化技术是本领域已知的,并且包括对上文所述转化技术的微小修改。本领域的技术人员可以选择利用瞬时转化来快速测定特定转基因的表达和/或功能,因为瞬时转化完成迅速,并且不需要像稳定转化技术那样多的资源(例如,为发育完整植物而进行的植物培养,为将转基因固定在基因组内而进行的植物自体受精或杂交,等)。
在一个实施方案中,供体多核苷酸可以引入到基本上任何植物中。有多种植物和植物细胞系统可以工程化以用于本公开的供体多核苷酸的位点特异性整合以及上述的各种转化方法。在一个实施方案中,用于工程化的靶植物和植物细胞包括,但不限于,那些单子叶植物和双子叶植物,例如作物,包括谷类作物(如小麦,玉米,大米,小米,大麦),水果作物(如番茄,苹果,梨,草莓,橘子),饲料作物(如苜蓿),根类蔬菜作物(例如,胡萝卜,马铃薯,糖用甜菜,山药),叶类蔬菜作物(如莴苣,菠菜);开花植物(例如,矮牵牛,月季,菊花),针叶树和松树(例如,松冷杉,云杉);在植物修复中使用的植物(例如,重金属积累植物);油料作物(例如,向日葵,油菜),以及用于实验目的的植物(例如,拟南芥)。
在其他实施方案中,为了基因组靶位点内的位点特异性靶向而将多核苷酸供体序列引入到植物细胞中。在这样的实施方案中,植物细胞可以是原生质体植物细胞。原生质体可以从各种类型的植物细胞产生。因此,本领域的普通技术人员可以利用不同的技术或方法学产生原生质体植物细胞。例如,下列文献中提供了原生质体的生成和产生:Green and Phillips,Crop Sc.,15(1975)417-421;Harms et al.Z.Pflanzenzuechtg.,77(1976)347-351;欧洲专利申请EP-0,160,390,Lowe and Smith(1985);EP-0,176,162,Cheng(1985);和EP-0,177,738,Close(1985);Cell Genetics in Higher Plants,Dudits et al.,(编辑),AkademiaiKiado,Budapest(1976)129-140,以及其中的参考文献;Harms,"Maize and Cereal Protoplasts-Facts and Perspectives,"Maize for Biological Research,W.F.Sheridan编辑(1982);Dale,收载于:Protoplasts(1983);Potrykus et al(编辑)Lecture Proceedings,ExperientiaSupplementum 46,Potrykus等编辑,Birkhauser,Basel(1983)31-41,以及其中的参考文献。从培养的原生质体再生植物在下列文献中有描述:Evans et al.(1983)“Protoplast Isolation and Culture,”Handbook of Plant Cell Cultures 1,124-176(MacMillan Publishing Co.,New York;Davey(1983)“Recent Developments in the Culture and Regeneration of Plant Protoplasts,”Protoplasts,第12-29页,(Birkhauser,Basel);Dale(1983)"Protoplast Culture and Plant Regeneration of Cereals and Other Recalcitrant Crops,"Protoplasts第31-41页,(Birkhauser,Basel);Binding(1985)"Regeneration of Plants,"Plant Protoplasts,第21-73页,(CRC Press,Boca Raton,FL)。
靶位点的选择、ZFP以及用于设计和构建融合蛋白(和编码它们的多核苷酸)的方法是本领域技术人员已知的,并且在下列文献中有详细描述:美国专利6,140,081;5,789,538;6,453,242;6,534,261;5,925,523;6,007,988;6,013,453;6,200,759;WO 95/19431;WO 96/06166;WO 98/53057;WO 98/54311;WO 00/27878;WO 01/60970;WO 01/88197;WO 02/099084;WO 98/53058;WO 98/53059;WO 98/53060;WO 02/016536和WO 03/016496。
在下面的实施方案中,包含一个或多个DNA结合序列的DNA结合结构域被锌指结合蛋白、大范围核酸酶结合蛋白、CRIPSR或TALEN结合蛋白所结合。
在某些实施方案中,本文描述的组合物和方法使用大范围核酸酶(归巢核酸内切酶)结合蛋白或大范围核酸酶DNA结合域来结合供体分子和/或结合细胞基因组中感兴趣的区域。天然存在的大范围核酸酶识别15-40个碱基对的切割位点,并且通常分为四个家族:LAGLIDADG家族、GIY-YIG家族、His-Cyst盒家族和HNH家族。示例性的归巢核酸内切酶包括I-SceI,I-CeuI,PI-PspI,PI-Sce,I-SceIV,I-CsmI,I-PanI,I-SceII,I-PpoI,I-SceIII,I-CreI,I-TevI,I-TevII和I-TevIII。它们的识别序列是已知的。另外参见美国专利5,420,032;美国专利6,833,252;Belfort et al.(1997)Nucleic Acids Res.25:3379–3388;Dujon et al.(1989)Gene 82:115–118;Perler et al.(1994)Nucleic Acids Res.22,1125–1127;Jasin(1996)Trends Genet.12:224–228;Gimble et al.(1996)J.Mol.Biol.263:163–180;Argast et al.(1998)J.Mol.Biol.280:345–353和New English Biolabs目录。
在某些实施方案中,本文描述的组合物和方法使用包含工程化的(非天然存在的)归巢核酸内切酶(大范围核酸酶)的核酸酶。归巢核酸内切酶和大范围核酸酶(例如I-SceI,I-CeuI,PI-PspI,PI-Sce,I-SceIV,I-CsmI,I-PanI,I-SceII,I-PpoI,I-SceIII,I-CreI,I-TevI,I-TevII和I-TevIII)的识别序列是已知的。另外参见美国专利5,420,032;美国专利6,833,252;Belfort et al.(1997)Nucleic Acids Res.25:3379–3388;Dujon et al.(1989)Gene 82:115–118;Perler et al.(1994)Nucleic Acids Res.22,1125–1127;Jasin(1996)Trends Genet.12:224–228;Gimble et al.(1996)J.Mol.Biol.263:163–180;Argast et al.(1998)J.Mol.Biol.280:345–353和New English Biolabs目录。此外,归巢核酸内切酶和大范围核酸酶的DNA结合特异性可以被工程化,以结合非天然的靶位点。参见例如,Chevalier et al.(2002)Molec.Cell 10:895-905;Epinat et al.(2003)Nucleic Acids Res.31:2952-2962;Ashworth et al.(2006)Nature441:656-659;Paques et al.(2007)Current Gene Therapy 7:49-66;美国专利公开No.20070117128。归巢核酸内切酶和大范围核酸酶的DNA结合域可以在以核酸酶为整体的环境中被改变(即,使得核酸酶包含关联切割结构域)。
在其它实施方案中,在本文所述方法和组合物中使用的一种或多种核酸酶之DNA结合域包括天然存在的或工程化的(非天然存在的)TAL效应子DNA结合域。参见例如,美国专利公开No.20110301073,其全部内容通过引用并入本文。已知黄单胞菌属的植物病原细菌会在重要农作物中导致许多疾病。黄单胞菌的致病性取决于保守的III型分泌(T3S)系统,其向植物细胞内注射25种以上的不同效应物蛋白。这些被注射蛋白包括转录激活子样(TAL)效应物,它们可模拟植物转录激活子并操纵植物转录组(参见Kay et al(2007)Science 318:648-651)。这些蛋白质包含DNA结合域和转录激活结构域。其中表征最充分的TAL效应物是来自野油菜叶斑病黄单胞菌(Xanthomonas campestgris pv.Vesicatoria)的AvrBs3(参见Bonas et al(1989)Mol Gen Genet 218:127-136和WO2010079430)。TAL效应物含有由串联重复构成的集中域,每一个重复含有大约34个氨基酸,它们对于这些蛋白质的DNA结合特异性是关键的。此外,它们含有核定位序列和酸性转录激活域(综述参见Schornack S,et al(2006)J Plant Physiol 163(3):256-272)。此外,在植物病原细菌青枯雷尔氏菌(Ralstonia solanacearum)中,已经发现两个基因,名为brg11和hpx17,与青枯雷尔氏菌生物变体1菌株GMI1000和生物变体4菌株RS1000中的黄单胞菌AvrBs3家族同源(参见Heuer et al(2007)Appl and Envir Micro 73(13):4379-4384)。这些基因在碱基序列上彼此98.9%相同,但在hpx17的重复结构域中有1575个碱基对缺失的差异。然而,这两种基因产物与黄单胞菌的AvrBs3家族蛋白的序列同一性均小于40%。参见例如,美国专利公开20110239315,20110145940和20110301073,其全部内容通过引用并入本文。
这些TAL效应物的特异性取决于在串联重复中发现的序列。重复序列包含大约102bp,并且各重复彼此之间通常91-100%同源(Bonas et al,同上)。重复的多态性通常位于位置12和13,并且位置12和13的高变双残基的同一性与TAL效应物靶序列中连续核苷酸的同一性似乎具有一对一的对应关系(参见Moscou and Bogdanove,(2009)Science 326:1501和Boch et al(2009)Science 326:1509-1512)。在实验上,这些TAL-效应物的DNA识别的自然编码已经被确定如下:位置12和13处的HD序列导致结合胞嘧啶(C),NG结合T,NI结合A、C、G或T,NN结合A或G,ING结合T。已有人将这些DNA结合重复组装成具有新的重复组合和数目的蛋白质,从而制造出能够与植物细胞中新的序列相互作用并激活非内源报告基因表达的人工转录因子(Boch et al,同上)。已有人将工程化的TAL蛋白与FokI切割半域连接,从而产生TAL效应物结构域核酸酶融合物(TALEN),其在酵母报告测定(基于质粒的靶标)中显示活性。参见例如美国专利公开20110301073;Christian et al((2010)<Genetics epub 10.1534/genetics.110.120717。
在其它实施方案中,核酸酶是包含CRISPR(成簇的、规律间隔的短回文重复(clustered regularly interspaced short palindromic repeats)/Cas(CRISPR相关的)核酸酶系统的系统。CRISPR/Cas是一种新近构建出来的核酸酶系统,基于一种能够用于基因组工程化的细菌系统。它是基于许多细菌和古细菌的适应性免疫应答的一部分。当病毒或质粒侵入细菌时,入侵者的DNA片段被“免疫”应答转化成CRISPR RNA(crRNA)。此crRNA随后借由一部分互补区域与另一种类型的称为tracrRNA的RNA相缔合,从而引导Cas9核酸酶到达与前间区序列邻近基序(protospacer adjacent motif)(PAM)NGG紧邻的靶DNA中的crRNA同源的区域。Cas9在被crRNA转录本内所含的一条20个核苷酸的引导序列所规定的位点处切割DNA,在DSB处产生平末端。Cas9的位点特异性DNA识别和切割既需要crRNA也需要tracrRNA。人们现已对这个系统进行了工程构建,使crRNA和tracrRNA能够组合成一个分子(“单引导RNA”),并且该单引导RNA的crRNA等效部分可以被工程化,以引导Cas9核酸酶靶向与PAM邻近的任何期望序列(参见Jinek et al(2012)Science337,p.816-821,Jinek et al,(2013),eLife 2:e00471,和David Segal,(2013)eLife 2:e00563)。因此,CRISPR/Cas系统可以被工程化,在基因组的期望靶标处创建DSB,并且该DSB的修复会受到使用修复抑制剂的影响,从而导致易错修复的增加。
在某些实施方案中,用于体内切割和/或靶向切割细胞基因组的一种或多种核酸酶的DNA结合域包括锌指蛋白。优选地,锌指蛋白是非天然存在的,因其工程化而结合所选靶位点。参见例如Beerli et al.(2002)Nature Biotechnol.20:135-141;Pabo et al.(2001)Ann.Rev.Biochem.70:313-340;Isalan et al.(2001)Nature Biotechnol.19:656-660;Segal et al.(2001)Curr.Opin.Biotechnol.12:632-637;Choo et al.(2000)Curr.Opin.Struct.Biol.10:411-416;美国专利6,453,242;6,534,261;6,599,692;6,503,717;6,689,558;7,030,215;6,794,136;7,067,317;7,262,054;7,070,934;7,361,635;7,253,273;和美国专利公开2005/0064474;2007/0218528;2005/0267061,所有文献的全部内容通过引用并入本文。
工程化的锌指结合域与天然存在的锌指蛋白相比可以具有新的结合特异性。工程化方法包括,但不仅限于,合理设计和各种类型选择。合理设计包括,例如,使用包括三联体(或四联体)核苷酸序列和各别锌指氨基酸序列的数据库,其中每个三联体或四联体核苷酸序列与结合该特定的三联体或四联体序列的一种或多种锌指氨基酸序列相关联。参见例如,共有的美国专利6,453,242和6534261,其全部内容通过引用并入本文。
示例性的选择方法,包括噬菌体展示和双杂交系统,在下列文献中有公开:美国专利5,789,538;5,925,523;6,007,988;6,013,453;6,410,248;6,140,466;6,200,759;and 6,242,568;以及WO 98/37186;WO 98/53057;WO 00/27878;WO 01/88197和GB 2,338,237。此外,对锌指结合域结合特异性的增强已经在例如共有的WO 02/077227中有描述。
此外,如这些和其它参考文献中公开的,锌指域和/或多指(multi-fingered)锌指蛋白可以使用任何合适的接头序列连接在一起,包括例如长度为5个或更多个氨基酸的接头。关于长度为6个或更多个氨基酸的示例性接头序列另外参见美国专利6,479,626;6,903,185;和7,153,949。本文所述的蛋白可以在该蛋白的各个锌指之间包含合适接头的任意组合。
本文描述可用于体内切割携带转基因的供体分子的组合物,特别是核酸酶,以及用于切割细胞基因组、从而使转基因以靶向的方式整合到基因组中的核酸酶。在某些实施方案中,一种或多种所述的核酸酶是天然存在的。在其它实施方案中,一种或多种所述的核酸酶是非天然存在的,因其在DNA结合域和/或切割结构域中被工程化。例如,天然存在的核酸酶的DNA结合域可以被改变,以结合选定的靶位点(即,大范围核酸酶已被工程化,从而结合与其关联结合位点不同的位点)。在其它实施方案中,核酸酶包括异源DNA结合和切割结构域(例如,锌指核酸酶;TAL效应物结构域DNA结合蛋白;具有异源切割结构域的大范围核酸酶DNA结合域)。
可以将任何合适的切割结构域与DNA结合域操作连接以形成核酸酶。例如,已有人将ZFP DNA结合域与核酸酶结构域融合产生了ZFN——一种功能实体,能够借助其工程化(ZFP)的DNA结合域识别其预期核酸靶标,并通过核酸酶活性导致DNA在临近ZFP结合位点处被切割。参见例如,Kim et al.(1996)Proc Natl AcadSci USA 93(3):1156-1160。更近来,ZFN已被用于在多种生物体中进行基因组修饰。参见例如,美国专利公开20030232410;20050208489;20050026157;20050064474;20060188987;20060063231;和国际公开WO07/014275。同样地,已有人将TALE DNA结合域与核酸酶域融合而产生了TALENS。参见例如,美国公开No.20110301073。
如上面指出的,切割域与DNA结合域可以是异源的,例如,锌指DNA结合域与来自核酸酶的切割域,或者TALEN DNA结合域与切割域,或者大范围核酸酶DNA结合域和来自不同核酸酶的切割域。异源切割域可以从某些核酸内切酶或核酸外切酶获得。可以产生切割域的示例性核酸内切酶包括,但不限于,某些限制性核酸内切酶和归巢核酸内切酶。参见例如,New English Biolabs2002-2003年产品目录,Beverly,MA;和Belfort et al.(1997)Nucleic Acids Res.25:3379-3388。其它可切割DNA的酶是已知的(例如,S1核酸酶;绿豆核酸酶;胰DNA酶I;微球菌核酸酶;酵母HO内切酶;另见Linn et al.(编)Nucleases,Cold Spring Harbor Laboratory Press,1993)。一种或多种这些酶(或其功能片段)可被用作切割域和切割半域的来源。
类似地,切割半域可以来源于任何切割活性需要二聚体化的核酸酶或其部分,如上文所述。一般地,如果融合蛋白包含切割半域,则需要两个融合蛋白才能切割。或者,可以使用包括两个切割半域的单个蛋白质。两个切割半域可以来自相同的核酸内切酶(或其功能片段),或者每个切割半域可以来自不同的核酸内切酶(或其功能片段)。另外,两个融合蛋白的靶位点优选地如此彼此相对布置,使得这两个融合蛋白与它们各自的靶位点结合后,各切割半域彼此的空间取向允许各切割半域形成一个功能性的切割域,例如通过二聚体化。因此,在某些实施方案中,靶位点的邻近边缘被间隔5-8个核苷酸或者15-18个核苷酸。然而,两个靶位点之间可以间隔任意整数的核苷酸或核苷酸对(例如,2-50个核苷酸对或以上)。一般地,切割位点位于靶位点之间。
限制性核酸内切酶(限制酶)存在于许多物种中,并能够序列特异性地结合DNA(在识别位点处),并在结合位点或附近切割DNA。某些限制性内切酶(例如IIS型)在远离识别位点的位点处切割DNA,且具有可分离的结合和切割域。例如,IIS型酶FokI催化DNA的双链切割,切割处在一条链上与其识别位点距离9个核苷酸,而在另一条链上与其识别位点距离13个核苷酸。参见例如,美国专利5356802;5436150和5487994;以及Li et al.(1992)Proc.Natl.Acad.Sci.USA 89:4275-4279;Li et al.(1993)Proc.Natl.Acad.Sci.USA 90:2764-2768;Kim et al.(1994a)Proc.Natl.Acad.Sci.USA 91:883-887;Kim et al.(1994b)J.Biol.Chem.269:31,978-31,982。因此,在一个实施方案中,融合蛋白包含来自至少一个IIS型限制性内切酶的切割域(或切割半域)和一个或多个锌指结合域,其可以被或者不被工程化。
一种切割域与其结合域分离的示例性IIS型限制酶是Fok I。这种酶作为二聚体发挥活性。Bitinaite et al.(1998)Proc.Natl.Acad.Sci.USA 95:10,570-10,575。因此,为本公开的目的,在公开的融合蛋白中使用的Fok I酶的一部分被认为是切割半域。因此,对于使用锌指-Fok I融合物的靶向双链切割和/或靶向DNA序列置换,可使用两个融合蛋白,每一个融合蛋白包含FokI切割半域,来重构有催化活性的切割域。或者,也可以使用包含锌指结合域和两个Fok I切割半域的单一多肽分子。使用锌指-Fok融合物改变靶切割和靶序列的参数在本公开的其它部分提供。
切割域和切割半域可以是蛋白质的任何保留切割活性、或者保留多聚体化(例如二聚体化)形成功能性切割域的能力的部分。
示例性的IIS型限制性内切酶在国际公开WO07/014275中有描述,其全全部内容并入本文。其它的限制性内切酶还包含可分离的结合和切割域,并且这些是本公开所构想的。参见例如,Roberts et al.(2003)Nucleic Acids Res.31:418-420。
在某些实施方案中,切割域包含一个或多个工程化的切割半域(也称作二聚体化结构域突变体),其可以最小化或者防止同二聚体化,如例如美国专利20050064474;20060188987;20070305346和20080131962中所述,其全部公开内容通过引用并入本文。Fok I的位置446,447,479,483,484,486,487,490,491,496,498,499,500,531,534,537,和538处的氨基酸残基均是影响Fok I切割半域二聚体化的靶点。
形成专性(obligate)异二聚体的FokⅠ的示例性工程化切割半域包括一对切割半域,其中第一个切割半域在Fok I的位置490和538处含有氨基酸残基突变,第二个切割半域在氨基酸残基486和499处含有突变。
因此,在一个实施方案中,490处的突变用Lys(K)代替Glu(E);538处的突变用Lys(K)代替Iso(I);486处的突变用Glu(E)代替Gln(Q);且位置499处的突变用Lys(K)代替Iso(I)。具体地,本文所述的工程化切割半域的制备是通过在一个切割半域中突变位置490(E→K)和538(I→K)以产生称为“E490K:I538K”的工程化切割半域,并通过在另一个切割半域中突变位置486(Q→E)和499(I→L)以产生称为“Q486E:I499L”的工程化切割半域。本文所述的工程化切割半域是专性异二聚体突变体,其中异常切割被最小化或者消除。参见例如,美国专利公开2008/0131962,其全部公开内容通过引用并入本文用于所有目的。在某些实施方案中,工程化的切割半域在位置486、499和496(相对于野生型FokI的编号)包括突变,例如在位置486用Glu(E)残基代替野生型的Gln(Q)残基、在位置499处用Leu(L)残基代替野生型的Iso(I)残基、和在位置496处用Asp(D)或Glu(E)残基代替野生型的Asn(N)残基(也分别被称作“ELD”和“ELE”域)的突变。在其它实施方案中,工程化的切割半域在位置490、538和537(相对于野生型FokI的编号)包含突变,例如在位置490用Lys(K)残基代替野生型的Glu(E)残基、在位置538处用Lys(K)残基代替野生型的Iso(I)残基、和在位置537处用Lys(K)残基或Arg(R)残基代替野生型的His(H)残基的突变(也分别被称作“KKK”和“KKR”结构域)。在其它实施方案中,工程化的切割半域包括在位置490和537(相对于野生型FokI的编号)的突变,例如在位置490用Lys(K)残基代替野生型的Glu(E)残基,和在位置537处用Lys(K)残基或Arg(R)残基代替野生型的His(H)残基的突变(也分别被称作“KIK”和“KIR”结构域)。(参见美国专利公开20110201055)。在其它实施方案中,工程化的切割半域包括“Sharkey”和/或“Sharkey’”突变(参见Guo et al,(2010)J.Mol.Biol.400(1):96-107)。
本文所述的工程化切割半域可以用任何合适的方法制备,例如,通过对野生型切割半域(Fok I)进行位点导向的突变,如美国专利公开20050064474;20080131962;和20110201055所述。
或者,核酸酶可以使用所谓的“分割酶”技术在体内核酸靶位点处组装(参见例如美国专利公开20090068164)。这样的分割酶的组分可以在不同的表达构建体上表达,或者可以连接在一个开放阅读框内,其中各个组分被分隔开,例如被自切割2A肽或IRES序列分隔开。组分可以是单个的锌指结合域或者大范围核酸酶核酸结合域的域。
在使用之前,可以筛选核酸酶的活性,例如基于酵母的染色体系统中筛选活性,如WO 2009/042163和20090068164所述。核酸酶表达构建体可以使用本领域已知的方法容易地设计。参见例如美国专利公开20030232410;20050208489;20050026157;20050064474;20060188987;20060063231;和国际公开WO 07/014275。核酸酶的表达可以处于组成型启动子或可诱导启动子的控制之下,例如半乳糖激酶启动子在棉子糖和/或半乳糖的存在下被激活(去阻遏),而在葡萄糖存在下被抑制。
在一个实施方案中,多核苷酸供体盒包含编码肽的序列。为了表达肽,编码肽序列的核苷酸序列通常被亚克隆到含有指导转录的启动子的表达载体中。合适的细菌和真核启动子在本领域是公知的,且在例如Sambrooket al.,Molecular Cloning,A Laboratory Manual(第2版,1989;第3版,2001);Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);和Current Protocols in Molecular Biology(Ausubelet al.,同上)中有描述。用于表达肽的细菌表达系统中可以在,例如,大肠杆菌、芽孢杆菌、和沙门氏菌中获得(Palvaet al.,Gene 22:229-235(1983))。用于此类表达系统的试剂盒可以商购获得。用于哺乳动物细胞、酵母和昆虫细胞的真核表达系统对于本领域技术人员而言是众所周知的,并且也可商购获得。
在一个实施方案中,多核苷酸供体盒包含基因表达盒,其包含转基因。该基因表达盒通常含有转录单元或表达盒,其包含在宿主细胞中,无论是原核生物还是真核生物,表达该核酸所需的全部附加元件。因此,典型的基因表达盒包含:启动子,可操作连接于例如编码蛋白质的核酸序列,以及信号(例如转录本高效多腺苷酸化所需的信号),转录终止子,核糖体结合位点,或翻译终止子。盒的其他元件可以包括,例如,增强子和异源剪接信号。
在一个实施方案中,基因表达盒还会在感兴趣的异源核苷酸序列的3'端包括在植物中有功能的转录和翻译终止区。该终止区可以是本公开实施方案的启动子核苷酸序列固有的,可以是感兴趣的DNA序列固有的,或者可以来自其他来源。方便的终止区可从根癌土壤杆菌的Ti质粒获得,例如章鱼碱合酶和胭脂碱合酶(nos)终止区(Depicker et al.,Mol.and Appl.Genet.1:561-573(1982)和Shaw et al.(1984)Nucleic Acids Research vol.12,No.20pp7831-7846(nos));另见Guerineau et al.Mol.Gen.Genet.262:141-144(1991);Proudfoot,Cell 64:671-674(1991);Sanfacon et al.Genes Dev.5:141-149(1991);Mogen et al.Plant Cell 2:1261-1272(1990);Munroe et al.Gene 91:151-158(1990);Ballas et al.Nucleic Acids Res.17:7891-7903(1989);Joshi et al.Nucleic Acid Res.15:9627-9639(1987)。
在其他实施方案中,基因表达盒可以另外含有5'前导序列。这样的前导序列可以起到增强翻译的作用。翻译前导序列是本领域已知的,并且包括例如,小核糖核酸病毒前导序列、EMCV前导序列(脑心肌炎5'非编码区)(Elroy-Stein et al.Proc.Nat.Acad.Sci.USA 86:6126-6130(1989));马铃薯Y病毒前导序列,例如,TEV前导序列(烟草蚀纹病毒)Carrington and Freed Journal of Virology,64:1590-1597(1990),MDMV前导序列(玉米矮花叶病毒),Allison et al.,Virology 154:9-20(1986);人免疫球蛋白重链结合蛋白(BiP),Macejak et al.Nature 353:90-94(1991);来自苜蓿花叶病毒外壳蛋白mRNA(AMV RNA 4)的非翻译前导序列,Jobling et al.Nature 325:622-625(1987);烟草花叶病毒前导序列(TMV),Gallie et al.(1989)Molecular Biology of RNA,237-256页;和玉米褪绿斑驳病毒前导序列(MCMV)Lommel et al.Virology81:382-385(1991)。另外参见Della-Cioppa et al.Plant Physiology 84:965-968(1987)。构建体还可以包含增强翻译和/或mRNA稳定性的序列,例如内含子。一种这样的内含子的实例是拟南芥组蛋白H3.III变异体的基因II的第一内含子。Chaubet et al.Journal of Molecular Biology,225:569-574(1992)。
在一个实施方案中,多核苷酸供体序列的基因表达盒包含启动子。用于指导肽编码核酸表达的启动子取决于具体的应用。例如,适合于宿主细胞的强组成型启动子通常用于表达和纯化蛋白质。优选的植物启动子的非限制性实例包括来自拟南芥泛素10(ubi-10)(Callis,et al.,1990,J.Biol.Chem.,265:12486-12493);根癌土壤杆菌甘露碱合酶(Δmas)(Petolinoet al.,美国专利6,730,824);和/或木薯叶脉花叶病毒(CsVMV)(Verdagueret al.,1996,Plant Molecular Biology 31:1129-1139)的启动子。
在本文所述的方法中,可以使用多种能够在植物中指导基因表达的启动子。这样的启动子可以选自组成型的、化学调节的、可诱导的、组织特异性的、和种子优先的启动子。
组成型启动子包括,例如,核心花椰菜花叶病毒35S启动子(Odell et al.(1985)Nature 313:810-812);水稻肌动蛋白启动子(McElroy et al.(1990)Plant Cell 2:163-171);玉米泛素启动子(美国专利号5510474;Christensen et al.(1989)Plant Mol.Biol.12:619-632和Christensen et al.(1992)Plant Mol.Biol.18:675-689);pEMU启动子(Last et al.(1991)Theor.Appl.Genet.81:581-588);ALS启动子(美国专利号5659026);玉米组蛋白启动子(Chaboutéet al.Plant Molecular Biology,8:179-191(1987));等等。
可用的植物相容性启动子的范围包括组织特异性和可诱导的启动子。可诱导的调控元件是能够响应诱导剂直接或间接激活一个或多个DNA序列或基因转录的元件。在没有诱导剂时,DNA序列或基因不会被转录。通常,特异性结合可诱导型调控元件来激活转录的蛋白因子以无活性的形式存在,然后被诱导剂直接或间接地转化为有活性的形式。诱导剂可以是化学介质例如蛋白质、代谢物、生长调节剂、除草剂或酚类化合物,或者是由热、冷、盐、或有毒成分直接施加的、或者通过病原体或致病剂如病毒的作用间接施加的生理胁迫。通常,特异性结合可诱导型调控元件来激活转录的蛋白因子以无活性的形式存在,然后被诱导剂直接或间接地转化为有活性的形式。诱导剂可以是化学介质例如蛋白质、代谢物、生长调节剂、除草剂或酚类化合物,或者是由热、冷、盐、或有毒成分直接施加,或者通过病原体或致病剂如病毒的作用间接施加的生理胁迫。将含有可诱导型调控元件的植物细胞暴露于诱导剂可以通过从外部施加诱导剂给细胞或植物来实现,例如通过喷雾、浇水、加热或类似的方法。
任何可诱导型启动子均可以在本公开的实施方案中。参见Ward et al.Plant Mol.Biol.22:361-366(1993)。示例性的可诱导型启动子包括蜕皮激素受体启动子(美国专利号6,504,082);来自ACE1系统的启动子,其响应铜(Mett et al.PNAS 90:4567-4571(1993));来自玉米的In2-1和In2-2基因,其响应苯磺酰胺除草剂安全剂(美国专利号5,364,780;Hershey et al.,Mol.Gen.Genetics 227:229-237(1991)和Gatz et al.,Mol.Gen.Genetics 243:32-38(1994));来自Tn10的Tet抑制子(Gatz et al.,Mol.Gen.Genet.227:229-237(1991);或者来自类固醇激素基因的启动子,该基因的转录活性受糖皮质激素的诱导(Schena et al.,Proc.Natl.Acad.Sci.U.S.A.88:10421(1991)和McNellis et al.,(1998)Plant J.14(2):247-257);玉米GST启动子,其被用作苗前除草剂的疏水亲电子化合物所激活(参见美国专利5,965,387和国际专利申请公开WO 93/001294);和烟草PR-1a启动子,其被水杨酸激活(参见Ono S,Kusama M,Ogura R,Hiratsuka K.,“Evaluation of the Use of the Tobacco PR-1a Promoter to Monitor Defense Gene Expression by the Luciferase Bioluminescence Reporter System,”BiosciBiotechnolBiochem.2011Sep23;75(9):1796-800)。其他受化学调节的感兴趣的启动子包括四环素诱导的和四环素阻遏的启动子(参见,例如,Gatz et al.,(1991)Mol.Gen.Genet.227:229-237,和美国专利号5,814,618和5,789,156)。
其它感兴趣的可调节启动子包括冷响应调节元件或热休克调节元件,其转录可分别响应于冷或热暴露转录而启动(Takahashi et al.,Plant Physiol.99:383-390,1992);醇脱氢酶基因的启动子(Gerlach et al.,PNAS USA79:2981-2985(1982);Walker et al.,PNAS 84(19):6624-6628(1987)),其可被无氧条件诱导;和来自豌豆rbcS基因或豌豆psaDb基因的光诱导型启动子(Yamamoto et al.(1997)Plant J.12(2):255-265);一种光诱导型调节元件(Feinbaum et al.,Mol.Gen.Genet.226:449,1991;Lam and Chua,Science248:471,1990;Matsuoka et al.(1993)Proc.Natl.Acad.Sci.USA90(20):9586-9590;Orozco et al.(1993)Plant Mol.Bio.23(6):1129-1138),植物激素可诱导的调节元件(Yamaguchi-Shinozaki et al.,Plant Mol.Biol.15:905,1990;Kares et al.,Plant Mol.Biol.15:225,1990),等等。可诱导的调控元件还可以是玉米In2-1或In2-2基因的启动子,其响应苯磺酰胺除草剂安全剂(Hershey et al.,Mol.Gen.Gene.227:229-237,1991;Gatz et al.,Mol.Gen.Genet.243:32-38,1994),和转座子Tn10的Tet抑制子(Gatz et al.,Mol.Gen.Genet.227:229-237,1991)。胁迫诱导型启动子包括盐/水胁迫诱导型启动子,例如P5CS(Zang et al.(1997)Plant Sciences 129:81-89);冷诱导型启动子,例如cor15a(Hajela et al.(1990)Plant Physiol.93:1246-1252),cor15b(Wilhelm et al.(1993)Plant MolBiol 23:1073-1077),wsc120(Ouellet et al.(1998)FEBS Lett.423-324-328),ci7(Kirch et al.(1997)Plant Mol Biol.33:897-909),ci21A(Schneider et al.(1997)Plant Physiol.113:335-45);干旱诱导型启动子,例如Trg-31(Chaudhary et al(1996)Plant Mol.Biol.30:1247-57),rd29(Kasuga et al.(1999)Nature Biotechnology 18:287-291);渗透压诱导型启动子,例如Rab17(Vilardell et al.(1991)Plant Mol.Biol.17:985-93)和osmotin(Raghothama et al.(1993)Plant MolBiol 23:1117-28);和热诱导型启动子,例如热休克蛋白(Barros et al.(1992)Plant Mol.19:665-75;Marrs et al.(1993)Dev.Genet.14:27-41),smHSP(Waters et al.(1996)J.Experimental Botany 47:325-338),和来自欧芹泛素启动子的热休克可诱导型元件(WO 03/102198)。其它胁迫诱导型启动子包括rip2(美国专利号5,332,808和美国公开2003/0217393)和rd29a(Yamaguchi-Shinozaki et al.(1993)Mol.Gen.Genetics 236:331-340)。某些启动子受创伤诱导,包括土壤杆菌pMAS启动子(Guevara-Garcia et al.(1993)Plant J.4(3):495-505)和土壤杆菌ORF13启动子(Hansen et al.,(1997)Mol.Gen.Genet.254(3):337-343)。
组织优先型启动子可以用于靶向增强具体植物组织内的转录和/或表达。当指优先表达时,意思是在具体植物组织中的表达水平高于其它植物组织。这些类型的启动子的实例包括种子优先表达,例如由菜豆蛋白启动子(Bustos et al.1989.The Plant Cell Vol.1,839-853),和玉米球蛋白-1基因(Belanger,et al.1991Genetics 129:863-972)提供的。对于双子叶植物,种子优先的启动子包括,但不仅限于,豆β-菜豆蛋白,油菜籽蛋白,β-伴大豆球蛋白,大豆凝集素,十字花科蛋白(cruciferin)等。对于单子叶植物,种子优先的启动子包括,但不仅限于,玉米15kDa玉米醇溶蛋白,22kDa玉米醇溶蛋白,27kDa玉米醇溶蛋白,γ玉米醇溶蛋白,糯(waxy),萎缩素(shrunken)1,萎缩素2,球蛋白1等。种子优先的启动子还包括那些指导基因主要在种子的特定组织中表达的启动子,例如γ玉米醇溶蛋白的胚乳层优先启动子,来自烟草的隐蔽启动子(cryptic promoter)(Fobert et al.1994.T-DNA tagging of a seed coat-specific cryptic promoter in tobacco.Plant J.4:567-577);来自玉米的P-基因启动子(Chopra et al.1996.Alleles of the maize P gene with distinct tissue specificities encode Myb-homologous proteins with C-terminal replacements.Plant Cell 7:1149-1158,勘误于Plant Cell.1997,1:109),来自玉米的球蛋白-1启动子(Belenger and Kriz.1991.Molecular basis for Allelic Polymorphism of the maize Globulin-1gene.Genetics 129:863-972),和指导在种皮或玉米粒外壳上表达的启动子,例如果皮特异性的谷氨酰胺合成酶启动子(Muhitch et al.,2002.Isolation of a Promoter Sequence From the Glutamine Synthetase1-2Gene Capable of Conferring Tissue-Specific Gene Expression in Transgenic Maize.Plant Science 163:865-872)。
基因表达盒可以包含5'前导序列。这样的前导序列可以起到增强翻译的作用。翻译前导序列是本领域已知的,并且包括例如,小核糖核酸病毒前导序列、EMCV前导序列(脑心肌炎5'非编码区)(Elroy-Stein et al.Proc.Nat.Acad.Sci.USA 86:6126-6130(1989));马铃薯Y病毒前导序列,例如,TEV前导序列(烟草蚀纹病毒)Carrington and Freed Journal of Virology,64:1590-1597(1990),MDMV前导序列(玉米矮花叶病毒),Allison et al.,Virology 154:9-20(1986);人免疫球蛋白重链结合蛋白(BiP),Macejak et al.Nature 353:90-94(1991);来自苜蓿花叶病毒外壳蛋白mRNA(AMV RNA 4)的非翻译前导序列,Jobling et al.Nature 325:622-625(1987);烟草花叶病毒前导序列(TMV),Gallie et al.(1989)Molecular Biology of RNA,237-256页;和玉米褪绿斑驳病毒前导序列(MCMV)Lommel et al.Virology 81:382-385(1991)。另外参见Della-Cioppa et al.Plant Physiology 84:965-968(1987)。
构建体还可以包含增强翻译和/或mRNA稳定性的序列,例如内含子。一种这样的内含子的实例是拟南芥组蛋白H3.III变体的基因II的第一内含子。Chaubet et al.Journal of Molecular Biology,225:569-574(1992)。
在那些期望将异源核苷酸序列的表达产物导向于特定细胞器,特别是质体、淀粉体,或者内质网,或者分泌到细胞表面或细胞外的场合,表达盒可以进一步包含转运肽的编码序列。这样的转运肽是本领域众所周知的,包括但不仅限于,如下蛋白的转运肽:酰基载体蛋白、RUBISCO的小亚基、植物EPSP合酶和向日葵(Helianthus annuus)(参见Lebrun et al.美国专利5,510,417)、玉米Brittle-1叶绿体转运肽(Nelson et al.Plant Physiol117(4):1235-1252(1998);Sullivan et al.Plant Cell 3(12):1337-48;Sullivan et al.,Planta(1995)196(3):477-84;Sullivan et al.,J.Biol.Chem.(1992)267(26):18999-9004)等。此外,嵌合叶绿体转运肽在本领域中是已知的,例如优化转运肽(参见,美国专利号5,510,471)。其它的叶绿体转运肽先前在美国专利5,717,084、5,728,925中已有描述。本领域的技术人员将容易地意识到可供用于将产物表达于特定的细胞器中的诸多选项。例如,大麦α淀粉酶序列经常被用于指导表达至内质网上。Rogers,J.Biol.Chem.260:3731-3738(1985)。
在一个实施方案中,多核苷酸供体盒包含转基因。本文的一些实施方案提供了构成基因表达盒的编码多肽的转基因。这样的转基因可以用于多种用途中的任何一种,以产生转基因植物。本文出于举例说明的目的提供了构成基因表达盒的转基因的具体实例,包括包含性状基因、RNAi基因或报告子/选择标记基因的基因表达盒。
在对基因进行工程化以用于在植物中表达时,预期宿主植物的密码子偏好性可以通过,例如,使用公众可得的DNA序列数据库发现有关植物基因组的密码子分布或者各种植物基因的蛋白编码区的信息,来加以确定。一旦在纸上或者芯片上设计出了优化的(例如,植物优化的)DNA序列,便可以在实验室中合成实际的DNA分子,其在序列上与设计的序列精确对应。这种合成的核酸分子可以被克隆或者以其他方式被精确地操纵,就好像它们是来自自然的或天然的来源一样。
在一个实施方案中,在本主题申请中公开了待表达的转基因。基因表达盒可以包括报告基因/选择标记基因、性状基因,或RNAi基因。下面进一步提供了选择标记基因、性状基因和RNAi基因的实例。在本申请中公开的方法的优势在于,它们提供了不依赖于转基因的蛋白产物的特定功能,或其它功能的种系转化子选择方法。
赋予害虫或疾病抗性的转基因或编码序列
(A)植物疾病抗性基因。植物防御经常通过植物中疾病抗性基因(R)的产物与病原体中相应的无毒性(Avr)基因的产物的特异相互作用而被激活。可以用克隆的抗性基因转化植物品种,从而工程构建对特定病原体株有抗性的植物。这些基因的实例包括:提供黄枝孢霉(Cladosporium fulvum)抗性的番茄Cf-9基因(Jones et al.,1994Science 266:789);,提供丁香假单胞杆菌番茄致病变种抗性的番茄Pto基因,其编码一种蛋白激酶(Martin et al.,1993Science262:1432),和提供丁香假单胞菌抗性的拟南芥RSSP2基因(Mindrinos et al.,1994Cell 78:1089)。
(B)苏云金芽孢杆菌蛋白质、其衍生物或以其为模本的人造多肽,例如Btδ-内毒素基因的多核苷酸序列(Geiser et al.,1986Gene 48:109)和植物杀虫(VIP)基因(见,例如,Estruch et al.(1996)Proc.Natl.Acad.Sci.93:5389-94)。此外,编码δ-内毒素基因的DNA分子可以从美国典型培养物保藏中心(Rockville,Md.)购得,ATCC登录号为40098,67136,31995和31998。
(C)植物凝集素,例如,多种君子兰(Clivia miniata)甘露糖结合性植物凝集素基因的核苷酸序列(Van Damme et al.,1994Plant Molec.Biol.24:825)。
(D)维生素结合蛋白质,例如亲和素及亲和素同源物,其可用作针对昆虫类害虫的杀幼虫剂。见美国专利No.5,659,026。
(E)酶抑制剂,例如蛋白酶抑制剂或淀粉酶抑制剂。这些基因的实例包括水稻半胱氨酸蛋白质酶抑制剂(Abe et al.,1987J.Biol.Chem.262:16793),烟草蛋白酶抑制剂I(Huub et al.,1993Plant Molec.Biol.21:985),和α-淀粉酶抑制剂(Sumitani et al.,1993Biosci.Biotech.Biochem.57:1243)。
(F)昆虫特异性激素或信息素,例如蜕皮激素和保幼激素或其变体、基于它们的模拟物,或其拮抗剂或激动剂,例如杆状病毒表达的克隆保幼激素酯酶,保幼激素的失活子(Hammock et al.,1990Nature 344:458)。
(G)昆虫特异性肽或神经肽,其在表达时会扰乱受影响的害虫的生理机能(J.Biol.Chem.269:9)。这些基因的实例包括昆虫利尿激素受体(Regan,1994),在太平洋折翅蠊(Diploptera punctata)中鉴定的咽侧体抑制素(allostatin)(Pratt,1989),和昆虫特异性麻痹神经毒素(美国专利No.5,266,361)。
(H)在自然界中由蛇、马蜂等产生的昆虫特异性毒液,例如蝎子昆虫毒性肽(Pang,1992Gene 116:165)。
(I)负责超富集单萜、倍半萜、甾体、异羟肟酸、苯丙烷衍生物或其它具有杀虫活性的非蛋白质分子的酶。
(J)参与生物活性分子修饰(包括翻译后修饰)的酶;例如糖酵解酶、蛋白质水解酶、脂肪分解酶、核酸酶、环化酶、转氨酶、酯酶、水解酶、磷酸酶、激酶、磷酸化酶、聚合酶、弹性蛋白酶、几丁质酶和葡聚糖酶,无论是天然的还是人造的。这些基因的实例包括马蹄莲(callas)基因(PCT公开的申请WO93/02197),几丁质酶编码序列(其可以从例如ATCC以登录号3999637和67152获得),烟草钩虫几丁质酶(Kramer et al.,1993Insect Molec.Biol.23:691),和欧芹ubi4-2多聚泛素基因(Kawalleck et al.,1993Plant Molec.Biol.21:673)。
(K)刺激信号转导的分子。这些分子的实例包括绿豆钙调蛋白cDNA克隆的核苷酸序列(Botella et al.,1994Plant Molec.Biol.24:757),和玉米钙调蛋白cDNA克隆的核苷酸序列(Griess et al.,1994Plant Physiol.104:1467)。
(L)疏水矩肽(hydrophobic moment peptide)。见例如美国专利Nos.5,659,026和5,607,914,后者教导了赋予疾病抗性的人造抗微生物肽。
(M)膜透性酶,通道形成剂或通道阻断剂,例如杀菌肽-β裂解肽类似物(Jaynes et al.,1993Plant Sci.89:43),其使转基因烟草植物对青枯病有抗性。
(N)病毒侵袭性蛋白质或由其衍生的复杂毒素。例如,在经转化的植物细胞中,病毒衣壳蛋白的积累可赋予针对该衣壳蛋白所来源的病毒以及相关病毒所致的病毒感染和/或疾病发展的抗性。已经给转化植物赋予了衣壳蛋白介导的,针对苜蓿花叶病毒、黄瓜花叶病毒、烟草条纹病毒、马铃薯X病毒、马铃薯Y病毒、烟草蚀纹病毒、烟草脆裂病毒和烟草花叶病毒的抗性。参见,例如,Beachy et al.(1990)Ann.Rev.Phytopathol.28:451。
(O)昆虫特异性抗体或由其衍生的免疫毒素。因此,靶向昆虫肠道关键代谢功能的抗体可以使受影响的酶失活,杀死昆虫。例如,Taylor等人(1994),在第七届国际分子植物-微生物相互作用研讨会(Seventh Int'l.Symposium on Molecular Plant Microbe Interactions)上的第497号摘要显示了转基因烟草中通过产生单链抗体片段的酶失活。
(P)病毒特异性抗体。见例如Tavladoraki et al.(1993)Nature 266:469,其显示了表达重组抗体基因的转基因植物被保护免于病毒攻击。
(Q)由病原体或寄生物自然产生的发育阻滞(developmental-arrestive)蛋白质。因此,真菌内切α-1,4-D多聚半乳糖醛酸酶通过溶解植物细胞壁的均聚-α-1,4-D-半乳糖醛酸而促进真菌定殖和植物营养素释放(Lamb et al.,1992)Bio/Technology 10:1436。Toubart等(1992Plant J.2:367)描述了豆类内切多聚半乳糖醛酸酶抑制蛋白的编码基因的克隆和表征。
(R)由植物自然产生的发育阻滞(developmental-arrestive)蛋白质,例如大麦核糖体失活基因,其提供了增加的针对真菌疾病的抗性(Longemann et al.,1992).Bio/Technology 10:3305。
(S)RNA干扰,其中用RNA分子抑制靶基因的表达。一个实施例中的RNA分子是部分或完全双链的,其触发沉默响应,导致dsRNA被切割成小的干扰RNA,它们随后被纳入到靶向复合体中,靶向复合体破坏同源的mRNA。见例如Fire等人,美国专利6,506,559;Graham等人,美国专利6,573,099。
赋予除草剂抗性的基因
(A)编码针对抑制生长点或分生组织的除草剂,例如咪唑啉酮类(imidazalinone)、磺酰苯胺类(sulfonanilide)或磺酰脲类除草剂的抗性或耐受性的基因。这类基因的实例编码一种突变ALS酶(Lee et al.,1988EMBOJ.7:1241),其也称AHAL酶(Miki et al.,1990Theor.Appl.Genet.80:449)。
(B)一种或多种额外的编码针对草甘膦抗性或耐受性的基因,所述抗性或耐受性是由突变体EPSP合酶和aroA基因赋予的,或者是通过一些基因如GAT(草甘膦乙酰转移酶)或GOX(草甘膦氧化酶)和其它膦酰基化合物,如草胺膦(pat和bar基因,DSM-2),和芳氧基苯氧基丙酸和环己二酮(ACC酶抑制剂编码基因)所致的代谢失活而获得的。见例如美国专利No.4,940,835,其公开了可赋予草甘膦抗性的EPSP形式的核苷酸序列。编码突变体aroA基因的DNA分子能够以ATCC登录号39256获得,突变体基因的核苷酸序列在美国专利No.4,769,061中公开。欧洲专利申请No.0 333 033和美国专利No.4,975,374公开了可赋予除草剂如L-草铵膦抗性的谷氨酰胺合酶基因的核苷酸序列。欧洲专利申请No.0 242 246提供了草铵膦乙酰转移酶基因的核苷酸序列。De Greef et al.(1989)Bio/Technology 7:61中描述了表达编码草铵膦乙酰转移酶活性的嵌合bar基因的转基因植物的产生。赋予针对芳氧基苯氧基丙酸和环己二酮如稀禾定和甲禾灵(haloxyfop)的抗性的示例性基因是Accl-S1,Accl-S2和Accl-S3基因,如Marshall et al.(1992)Theor.Appl.Genet.83:435所述。
(C)编码针对可抑制光合作用的除草剂例如三嗪(psbA和gs+基因)和苄腈(腈水解酶基因)的抗性的基因。Przibilla et al.(1991)Plant Cell 3:169描述了使用编码突变体psbA基因的质粒转化衣藻。在美国专利No.4,810,648中公开了腈水解酶基因的核苷酸序列,含有这些基因的DNA分子可以通过ATCC登录号53435、67441和67442获得。Hayes et al.(1992)Biochem.J.285:173中描述了编码谷胱甘肽S-转移酶的DNA的克隆和表达。
(D)编码针对可结合羟基苯基丙酮酸二加氧酶(HPPD)的除草剂的抗性基因,HPPD是催化对-羟基苯基丙酮酸(HPP)转化形成尿黑酸的反应的酶。这包括例如异噁唑(EP418175,EP470856,EP487352,EP527036,EP560482,EP682659,美国专利No.5,424,276),特别是异噁唑草酮,其是玉米的选择性除草剂,二酮腈(diketonitrile)(EP496630,EP496631),特别是2-氰基-3-环丙基-1-(2-SO2CH3-4-CF3苯基)丙烷-1,3-二酮和2-氰基-3-环丙基-1-(2-SO2CH3-4-2,3Cl2苯基)丙烷-1,3-二酮,三酮类(EP625505,EP625508,美国专利No.5,506,195),特别是磺草酮、和pyrazolinate等除草剂。在植物中产生过量HPPD的基因能够提供针对这些除草剂的耐受性或抗性,包括例如美国专利Nos.6,268,549和6,245,968和美国专利申请公开No.20030066102中描述的基因。
(E)编码针对苯氧基生长素除草剂,如2,4-二氯苯氧基乙酸(2,4-D)的抗性或耐受性的基因,其也可以赋予针对芳氧基苯氧基丙酸类(AOPP)除草剂的抗性或耐受性。这些基因的实例包括α-酮戊二酸依赖性的双加氧酶(aad-1)基因,如美国专利No.7,838,733所述。
(F)编码针对苯氧基生长素除草剂如2,4-二氯苯氧基乙酸(2,4-D)的抗性或耐受性的基因,其也可以赋予针对吡啶基氧基生长素除草剂,如氟草烟或绿草定的抗性或耐受性。这些基因的实例包括α-酮戊二酸依赖性的双加氧酶(aad-12)基因,如WO2007/053482-A2所述。
(G)编码针对麦草畏的抗性或耐受性的基因(见例如美国专利公开No.20030135879)。
(H)编码针对抑制原卟啉原氧化酶(PPO)的除草剂的抗性或耐受性的基因(见美国专利No.5,767,373)。
(I)提供针对可结合光系统II反应中心(PS II)核心蛋白质的三嗪除草剂(例如莠去津)和尿素衍生物(如敌草隆)除草剂的抗性或耐受性的基因。见Brussian et al.,(1989)EMBO J.1989,8(4):1237-1245。
可赋予或贡献数量叠加性状(Value Added Trait)的基因
(A)修饰的脂肪酸代谢,例如通过用反义基因或硬脂酰-ACP去饱和酶转化玉米或芸苔属植物从而增加植物的硬脂酸含量(Knultzon et al.,1992)Proc.Nat.Acad.Sci.USA 89:2624。
(B)降低的植酸含量
(1)引入植酸酶编码基因,如黑曲霉植酸酶基因(Van Hartingsveldt et al.,1993Gene 127:87),提高植酸降解,向被转化植物添加更多游离磷酸盐。
(2)可引入降低植酸含量的基因。在玉米中,这可以通过,例如,克隆然后重新导入如下所述的单个等位基因的相关DNA来实现:该单个等位基因导致以植酸水平低为特征的玉米突变体的原因(Raboy et al.,1990Maydica 35:383)。
(C)改良的碳水化合物组成,例如通过用编码改变淀粉的分支模式的酶的基因转化植物而实现。这些酶的实例包括,粘液链球菌(Streptococcus mucus)果糖基转移酶基因(Shiroza et al.,1988)J.Bacteriol.170:810,枯草芽孢杆菌果聚糖蔗糖酶基因(Steinmetz et al.,1985Mol.Gen.Genel.200:220),地衣芽孢杆菌α-淀粉酶(Pen et al.,1992Bio/Technology 10:292),番茄转化酶基因(Elliot et al.,1993),大麦淀粉酶基因(Sogaard et al.,1993J.Biol.Chem.268:22480),和玉米胚乳淀粉分支酶II(Fisher et al.,1993Plant Physiol.102:10450)。
在随后的实施方案中,转基因包含报告基因。在各种不同的实施方案中,报告基因选自下组:yfp基因,gus基因,rfp基因,gfp基因,卡那霉素抗性基因,aad-1基因,aad-12基因,pat基因,和草甘膦耐受基因。用于选择被转化细胞或组织或植物部分或植物的报告基因或标记基因可以包含在转化载体中。选择标记的实例包括那些可以赋予对抗代谢物例如除草剂或抗生素的抗性的标记,例如,二氢叶酸还原酶,其赋予对氨甲蝶呤的抗性(Reiss,Plant Physiol.(Life Sci.Adv.)13:143-149,1994;另见Herrera Estrella et al.,Nature303:209-213,1983;Meijer et al.,Plant Mol.Biol.16:807-820,1991);新霉素磷酸转移酶,其赋予对氨基糖苷类新霉素、卡那霉素和巴龙霉素的抗性(Herrera-Estrella,EMBO J.2:987-995,1983和Fraley et al.Proc.Natl.Acad.Sci USA 80:4803(1983));潮霉素磷酸转移酶,其赋予对潮霉素的抗性(Marsh,Gene 32:481-485,1984;另见Waldron et al.,Plant Mol.Biol.5:103-108,1985;Zhijian et al.,Plant Science 108:219-227,1995);trpB,其允许细胞利用吲哚代替色氨酸;hisD,其允许细胞利用组氨醇代替组氨酸(Hartman,Proc.Natl.Acad.Sci.,USA 85:8047,1988);甘露糖-6-磷酸异构酶,其允许细胞利用甘露糖(WO 94/20627);鸟氨酸脱羧酶,其赋予对鸟氨酸脱羧酶抑制剂,2-(二氟甲基)-DL-鸟氨酸(DFMO)的抗性(McConlogue,1987,收载于:Current Communications in Molecular Biology,Cold Spring Harbor Laboratory编辑);和来自土曲霉的脱氨酶,其赋予对杀稻瘟菌素S的抗性(Tamura,Biosci.Biotechnol.Biochem.59:2336-2338,1995)。
其他的选择标记包括,例如,突变的乙酰乳酸合酶,其赋予咪唑啉酮或磺酰脲抗性(Lee et al.,EMBO J.7:1241-1248,1988),突变的psbA,其赋予对阿特拉津的抗性(Smeda et al.,Plant Physiol.103:911-917,1993),或突变的原卟啉原氧化酶(参见美国专利5,767,373);或其它可赋予对除草剂如草铵膦的抗性的标记物。合适的选择标记基因的实例包括,但不仅限于,编码对如下物质的抗性的基因:氯霉素(Herrera Estrella et al.,EMBO J.2:987-992,1983);链霉素(Jones et al.,Mol.Gen.Genet.210:86-91,1987);壮观霉素(Bretagne-Sagnard et al.,Transgenic Res.5:131-137,1996);博莱霉素(Hille et al.,Plant Mol.Biol.7:171-176,1990);磺酰胺(Guerineau et al.,Plant Mol.Biol.15:127-136,1990);溴苯腈(Stalker et al.,Science 242:419-423,1988);草甘膦(Shaw et al.,Science 233:478-481,1986);膦丝菌素(DeBlock et al.,EMBO J.6:2513-2518,1987),和类似物。
使用选择性基因的一个选项是草铵膦抗性编码DNA,并且在一个实施方案中可以是在木薯叶脉花叶病毒启动子控制下的膦丝菌素乙酰转移酶(pat)、玉米优化的pat基因、或bar基因。这些基因赋予对双丙氨膦的抗性。参见Wohlleben et al.,(1988)Gene 70:25-37;Gordon-Kamm et al.,Plant Cell2:603;1990;Uchimiya et al.,BioTechnology 11:835,1993;White et al.,Nucl.Acids Res.18:1062,1990;Spencer et al.,Theor.Appl.Genet.79:625-631,1990;和Anzai et al.,Mol.Gen.Gen.219:492,1989)。Pat基因的一个版本是玉米优化的pat基因,如美国专利6,096,947所述。
此外,可以使用能易化含有编码标记的多核苷酸的植物细胞的鉴定的标记。可评分或可筛选的标记是有用的,其中当该序列存在时会产生可以测量的产物,并可以在不破坏植物细胞的条件下生成产物。实例包括β-葡糖醛酸酶或uidA基因(GUS),其编码各种发色底物已知的酶(例如,美国专利5,268,463和5,599,670);氯霉素乙酰转移酶(Jefferson et al.The EMBO Journal vol.6No.13,3901-3907页);和碱性磷酸酶。在优选的实施方案中,所使用的标记物是β-胡萝卜素或维生素A原(Ye et al,Science 287:303-305-(2000))。该基因已经被用于提高水稻的营养,但在这种情况下它被用作可筛选标记,并当该基因与感兴趣的基因的连锁存可以通过所提供的而金黄色被检测出来。与利用该基因为植物贡献营养的情况不同的是,少量的蛋白即足以达到目的。其它的可筛选标记包括一般意义上的花青素/类黄酮基因(参见Taylor and Briggs,The Plant Cell(1990)2:115-127的讨论部分),其包括例如一种R-基因座基因,其编码的产物可以调节植物组织中花青素(红色)的产生(Dellaporta等人,收载于Chromosome Structure and Function,Kluwer Academic Publishers,Appels and Gustafson编辑,263-282页(1988));控制类黄酮色素生物合成的基因,例如玉米C1基因(Kao et al.,Plant Cell(1996)8:1171-1179;Scheffler et al.Mol.Gen.Genet.(1994)242:40-48)和玉米C2基因(Wienand et al.,Mol.Gen.Genet.(1986)203:202-207);B基因(Chandler et al.,Plant Cell(1989)1:1175-1183),p1基因(Grotewold et al,Proc.Natl.Acad.Sci USA(1991)88:4587-4591;Grotewold et al.,Cell(1994)76:543-553;Sidorenko et al.,Plant Mol.Biol.(1999)39:11-19);青铜色基因座基因(Ralston et al.,Genetics(1988)119:185-197;Nash et al.,Plant Cell(1990)2(11):1039-1049),等等。
合适标记物的进一步的实例包括青色荧光蛋白(CYP)基因(Bolte et al.(2004)J.Cell Science 117:943-54and Kato et al.(2002)Plant Physiol 129:913-42),黄色荧光蛋白基因(Evrogen的PHIYFPTM;见Bolte et al.(2004)J.Cell Science 117:943-54),lux基因,其编码荧光素酶,其存在可以使用例如X-射线胶片、闪烁计数、荧光光度法、低光摄像机、光子计数照相机或多孔发光测定(multiwellluminometry)来检测(Teeri et al.(1989)EMBO J.8:343);绿色荧光蛋白(GFP)基因(Sheen et al.,Plant J.(1995)8(5):777-84)和DsRed2,其中被标记基因转化的植物细胞呈红色,因此可以视觉选择(Dietrich et al.(2002)Biotechniques 2(2):286-293)。其它的实例包括β-内酰胺酶基因(Sutcliffe,Proc.Nat'l.Acad.Sci.U.S.A.(1978)75:3737),其编码具有多种已知的发色底物的酶(例如,PADAC,发色头孢菌素);xylE基因(Zukowsky et al.,Proc.Nat'l.Acad.Sci.U.S.A.(1983)80:1101),其编码儿茶酚双加氧酶,后者能够转换成生色儿茶酚;α-淀粉酶基因(Ikuta et al.,Biotech.(1990)8:241);和酪氨酸酶基因(Katz et al.,J.Gen.Microbiol.(1983)129:2703),其编码的酶能够将酪氨酸氧化成DOPA和多巴醌,其进而缩合(condense)形成容易检测的化合物黑色素。显然,有许多这样的标记可用,并且是本领域技术人员已知的。
术语“百分比同一性”(或“%同一性”),如本领域中已知的,是指如通过比较序列加以确定的两个或多个多肽序列或者两个或多个多核苷酸序列之间的关系。在本领域中,“同一性”也指多肽或多核苷酸序列之间的序列相关的程度,例如可以通过这些序列的字符串之间的匹配度来确定。“同一性”和“相似度”可以通过已知的方法容易地计算,包括但不仅限于在下述文献中公开的那些:1.)Computational Molecular Biology(Lesk,A.M.,Ed.)Oxford University:NY(1988);2.)Biocomputing:Informatics and Genome Projects(Smith,D.W.,Ed.)Academic:NY(1993);3.)Computer Analysis of Sequence Data,Part I(Griffin,A.M.,and Griffin,H.G.,Eds.)Humania:NJ(1994);4.)Sequence Analysis in Molecular Biology(von Heinje,G.,Ed.)Academic(1987);and 5.)Sequence Analysis Primer(Gribskov,M.and Devereux,J.,Eds.)Stockton:NY(1991)。
用于确定核酸和氨基酸序列同一性的技术是本领域公知的。通常,这样的技术包括确定某个基因mRNA的核苷酸序列和/或确定由其编码的氨基酸序列,并将这些序列与第二个核苷酸或氨基酸序列进行比较。基因组序列也可以通过这种方式加以确定和比较。一般地,同一性是分别指两个多核苷酸之间的精确核苷酸-核苷酸对应关系或两个多肽序列的精确氨基酸-氨基酸对应关系。两个或更多个序列(多核苷酸或氨基酸)可以通过确定它们的百分比同一性进行比较。两个序列的百分比同一性,不论是核酸还是氨基酸序列,是两个对齐的序列之间精确匹配的数目除以较短序列的长度并乘以100。参见Russell,R.,and Barton,G.,“Structural Features can be Unconserved in Proteins with Similar Folds,”J.Mol.Biol.244,332-350(1994),第337页,其全部内容通过引用并入本文。
此外,用于确定同一性和相似度的方法被编码在公众可得的计算机程序中。序列比对和百分比同一性的计算可以使用例如Vector 软件包(Invitrogen,Carlsbad,CA)的AlignX程序或LASERGENE生物信息学计算软件包(DNASTAR Inc.,Madison,WI)的MegAlignTM程序进行。序列的多重比对可以使用“Clustal比对方法”进行,其包括该算法的多种变化形式,包括相应于标记为Clustal V的比对方法的“Clustal V比对方法”(如Higgins and Sharp,CABIOS.5:151-153(1989);Higgins,D.G.et al.,Comput.Appl.Biosci.,8:189-191(1992)公开),并可在LASERGENE生物信息学计算软件包(DNASTAR Inc.)的MegAlignTM程序中找到。为了多重比对,默认值相应于GAP PENALTY=10和GAP LENGTH PENALTY=10。使用Clustal方法的蛋白质序列两两比对和百分比同一性计算的默认参数是KTUPLE=1,GAP罚分=3,WINDOW=5和DIAGONALS SAVED=5。对于核酸,这些参数是KTUPLE=2,GAP PENALTY=5,WINDOW=4和DIAGONALS SAVED=4。在使用Clustal V程序进行序列比对之后,可以通过在相同程序中检视“序列距离”表获知“百分比同一性”。此外,有“Clustal W比对方法”,其对应于标记为Clustal W的比对方法(如Higgins and Sharp,CABIOS.5:151-153(1989);Higgins,D.G.et al.,Comput.Appl.Biosci.8:189-191(1992))所述,并可在LASERGENE生物信息学计算软件包(DNASTAR Inc.)的MegAlignTM v6.1程序中找到。对于多重比对的默认参数是(GAP PENALTY=10,GAP LENGTH PENALTY=0.2,Delay DivergenSeqs(%)=30,DNA Transition Weight=0.5,Protein Weight Matrix=Gonnet Series,DNA Weight Matrix=IUB)。在使用Clustal W程序对序列进行比对之后,可以通过在相同程序中检视“序列距离”表获知“百分比同一性”。
本领域的技术人员熟知,可以使用许多不同水平的序列同一性从其它物种鉴定多肽,这些其他物种中所述多肽具有相同或相似的功能或活性。有用的百分比同一性的实例包括,但不仅限于:55%,60%,65%,70%,75%,80%,85%,90%,或95%;或者55%至100%的任意整数百分数均可用于描述本公开的实施方案,例如55%,56%,57%,58%,59%,60%,61%,62%,63%,64%,65%,66%,67%,68%,69%,70%,71%,72%,73%,74%,75%,76%,77%,78%,79%,80%,81%,82%,83%,84%,85%,86%,87%,88%,89%,90%,91%,92%,93%,94%,95%,96%,97%,98%或99%。合适的核酸片段不仅具有上述的同源性还通常编码具有至少50个氨基酸,优选地至少100个氨基酸,更优选地至少150个氨基酸,更加优选地至少200个氨基酸,最优选地至少250个氨基酸的多肽。
术语“序列分析软件”是指可用于分析核苷酸或氨基酸序列的任何计算算法或软件程序。“序列分析软件”可以商业获得或者独立开发。典型的序列分析软件包括,但不仅限于:1)GCG程序软件包(Wisconsin Package Version9.0,Genetics Computer Group(GCG),Madison,WI);2.)BLASTP,BLASTN,BLASTX(Altschul et al.,J.Mol.Biol.,215:403-410(1990));3.)DNASTAR(DNASTAR,Inc.Madison,WI);4.)Sequencher(Gene Codes Corporation,Ann Arbor,MI);和5.)整合了Smith-Waterman算法的FASTA程序(W.R.Pearson,Comput.Methods Genome Res.,[Proc.Int.Symp.](1994),会议日期1992,111-20.编辑:Suhai,Sandor.Plenum:New York,NY)。在本申请的上下文中应当理解,在使用序列分析软件进行分析时,除非另外指出,否则分析结果是基于所提到的程序的“默认值”。如本文所使用的,“默认值”意思是软件在首次初始化时最初加载的任何一组设定值或参数。
当指称杂交技术时,已知核苷酸序列的全部或一部分可以用作探针,与来自所选生物体的克隆基因组DNA片段或cDNA片段群体(即,基因组或cDNA文库)中存在的其他对应的核苷酸序列选择性杂交。杂交探针可以是基因组DNA片段、质粒DNA片段、cDNA片段、RNA片段、PCR扩增的DNA片段、寡核苷酸或其它多核苷酸,并可以用可检测的基团如32P或任何其它可检测标记来标记。因此,杂交探针可以通过标记基于本公开实施方案的DNA序列的合成寡核苷酸而制得。用于制备杂交探针和构建cDNA和基因组文库的方法在本领域是公知的,并已被公开(Sambrook et al.,1989)。
本公开实施方案的核酸探针和引物在严格条件下与靶DNA序列杂交。可以使用任何常规的核酸杂交或扩增方法鉴定样品中来自转基因事件的DNA的存在。核酸分子或其片段在某些环境下能够与其它核酸分子特异性杂交。如本文所使用的,如果两个分子能够形成反向平行的双链核酸结构,则两个核酸分子被认为是能够彼此特异性杂交的。如果两个核酸分子表现出完全的互补性,则一个核酸分子称为另一个核酸分子的“互补物”。如本文所使用的,当其中一个分子的每一个核苷酸均与另一个的核苷酸互补时,则称这些分子显示“完全的互补性”。显示完全互补性的分子一般会以足够的稳定性彼此杂交,从而允许它们在常规的“高严格性”条件下保持彼此退火。常规的高严格性条件如Sambrook et al.,1989所述。
如果两个分子彼此杂交的稳定性足以使它们至少在常规的“低严格性”条件下保持彼此退火,则称它们显示“最小互补性”。常规的低严格性条件如Sambrook et al.,1989所述。为了让一个核酸分子能够用作引物或探针,它仅需要显示最小序列互补性,以便能够在所采用的具体溶剂和盐浓度下形成稳定的双链结构。
影响杂交严格性的因素是本领域技术人员众所周知的,并且包括,但不仅限于,温度、pH、离子强度和有机溶剂如甲酰胺和二甲亚砜的浓度。如本领域的技术人员所知的,杂交严格性随着温度升高、离子强度降低和溶剂浓度降低而增加。
术语“严格条件”或“严格性条件”是参照核酸探针与靶核酸(即,感兴趣的具体核酸序列)按照如Sambrooket al.,1989在9.52-9.55中所讨论的具体杂交程序进行的杂交而功能性定义的。另见Sambrooket al.,1989的9.47-9.52和9.56-9.58。
通常,严格条件是指盐浓度小于大约1.5M Na+离子,通常为大约0.01-1.0M Na+离子浓度(或其它盐),pH 7.0-8.3,温度为至少大约30℃(对于短探针,例如10-50个核苷酸)和至少大约60℃(对于长探针,例如大于50个核苷酸)。严格条件还可以通过添加去稳定剂如甲酰胺实现。示例性的低严格性条件包括用30-35%甲酰胺、1.0M NaCl、0.1%SDS(十二烷基磺酸钠)的缓冲溶液在37℃杂交,在1X至2X SSC(20X SSC=3.0M NaCl/0.3M柠檬酸三钠)中在50-55℃下清洗。示例性的中等严格性条件包括在40-45%甲酰胺、1.0M NaCl、0.1%SDS中在37℃杂交,和在0.5X至1X SSC中在55-60℃下清洗。示例性的高严格性条件包括在50%甲酰胺、1.0M NaCl、0.1%SDS中在37℃杂交,和在0.1X SSC中在60-65℃下清洗。
特异性通常依赖杂交后清洗而变化,关键的因素是最终清洗溶液的离子强度和温度。对于DNA-DNA杂交,Tm可以由公式Tm=81.5℃+16.6(logM)+0.41(%GC)-0.61(%form.)-500/L近似出来,其中M是单价阳离子的摩尔浓度,%GC是DNA中鸟嘌呤和胞嘧啶核苷酸的百分比,%form.是杂交溶液中甲酰胺的百分比,L是以碱基对计的杂交体长度(Meinkoth和Wahl,1984)。Tm是(在限定的离子强度和pH下)50%的互补靶序列与完美匹配的探针杂交的温度。每1%错配Tm降低大约1℃;因此,可以调节Tm、杂交、和/或清洗条件以令具有期望杂交同一性的序列发生杂交。例如,如果寻求具有90%同一性的序列,可以把Tm降低10℃。一般地,严格条件被选择为比限定的离子强度和pH下特定序列及其互补序列的热熔点(Tm)低大约5℃。然而,极严格条件可以在比热熔点(Tm)低1、2、3、或4℃下进行杂交和/或清洗;中等严格条件可以在比热熔点(Tm)低6,7,8,9,或10℃下进行杂交和/或清洗;低严格条件可以在比热熔点(Tm)低11-20℃下进行杂交和/或清洗。普通技术人员会理解,使用该公式、杂交和清洗组合物以及期望的Tm,即隐含描述了杂交和/或清洗溶液之严格性的变化。如果期望程度的错配导致Tm低于45℃(水溶液)或32℃(甲酰胺溶液),则优选地增加SSC浓度以便能够使用更高的温度。关于核酸杂交的详细指导参见(1997)Ausubelet al,Short Protocols in Molecular Biology,第2-40页,第三版(1997)和Sambrook et al.(1989)。
在本公开的另一个实施方案中,公开了用于将多核苷酸供体盒靶向整合在植物细胞基因组中的方法。在某些实施方案中,表达位点特异性DNA结合核酸酶,该核酸酶包含至少一个DNA结合域和至少一个核酸酶域,其中该至少一个DNA结合域与植物细胞基因组内的靶位点结合。在其它实施方案中,植物细胞与多核苷酸供体序列接触。在进一步的实施方案中,植物细胞基因组内的靶位点被位点特异性DNA结合核酸酶切割。在另外一个实施方案中,多核苷酸供体盒被整合到植物细胞基因组内的靶位点中。
在一个实施方案中,公开了通过同源性指导的修复机制将多核苷酸供体盒靶向整合在植物细胞的基因组中。在另一个实施方案中,公开了通过非同源末端连接指导的修复机制将多核苷酸供体盒靶向整合在植物细胞的基因组中。
本文公开的供体分子通过靶向、不依赖同源性的方法整合到细胞基因组中。这种靶向整合使用核酸酶,例如DNA结合域(例如,经工程化而结合预定切割位点处或其附近的靶位点的锌指结合域或TAL效应物结构域)和核酸酶结构域(例如,切割域或切割半域)的融合物,在预定位置(或多个位置)切割基因组。在某些实施方案中,在细胞中表达两种融合蛋白,每种融合蛋白包括一个DNA结合域和一个切割半域,并与靶位点结合,其中各靶位点以一定的方式并置,导致重构出一个功能性的切割域,并在靶位点附近切割DNA。在一个实施方案中,切割发生在两个DNA结合域的各靶位点之间。DNA结合域之一或二者可以被工程化。另外参见美国专利7,888,121;美国专利公开20050064474和国际专利公开WO05/084190、WO05/014791和WO03/080809。
如本文所述的核酸酶可以作为多肽和/或多核苷酸被引入。例如,可以引入两个多核苷酸到细胞中,其中每一个多核苷酸包含编码上述多肽的序列,并且当各多肽被表达并且各自与其靶序列结合时,在靶序列处或其附近发生切割。或者,将包含编码两个融合多肽的序列的单个多核苷酸引入到细胞中。多核苷酸可以是DNA、RNA或者DNA和/或RNA的任何经修饰形式或类似物。
在感兴趣区域内引入双链断裂之后,在如本文所述地将双链供体分子线性化之后,转基因通过非同源性依赖的方法(例如,非同源末端连接(NHEJ))以靶向方式被整合到感兴趣的区域内。双链供体优选地用核酸酶体内线性化,例如用与在基因组中引入双链断裂所用的核酸酶相同或不同的一种或多种核酸酶体内线性化。染色体和供体在细胞中的同步切割可以限制供体DNA降解(与先线性化供体然后引入到细胞内相比)。用于线性化供体的核酸酶靶位点优选地不破坏转基因序列。
转基因可以沿着核酸酶突出端简单连接所预期的方向(称作“正向”或“AB”方向)引入到基因组中,或者沿着可选择的方向(称作“反向”或“BA”方向)。在某些实施方案中,转基因在供体和染色体突出端准确连接之后整合。在其他实施方案中,转基因沿着BA或AB方向的整合导致数个核苷酸缺失。
IV.用于检测供体多核苷酸位点特异性整合的测定法
在一个实施方案中,对扩增反应进行定量。在其它实施方案中,对扩增反应进行检测。在各种的实施方案中,检测可以包括在琼脂糖或丙烯酰胺凝胶上可视化、扩增子测序、或使用标识概貌(signature profile),其中标识概貌(signature profile)从温度或荧光标识分布中选出。
本公开实施方案的核酸分子或其节段可以用作PCR扩增的引物。在进行PCR扩增时,可以容忍引物和模板之间有一定程度的错配。因此,示例引物的突变、缺失、和插入(特别是在5'或3'端的核苷酸添加)在本公开的范围之内。突变、插入和缺失可以通过本领域普通技术人员已知的方法在给定的引物中产生。
另一种检测实例是焦磷酸测序技术,如Winge(Innov.Pharma.Tech.00:18-24,2000)所述。在这种方法中,设计寡核苷酸与相邻的基因组DNA和插入DNA的接点重叠。该寡核苷酸与来自感兴趣区域的单链PCR产物杂交(一个引物在插入序列中,另一个在侧翼基因组序列中),并在DNA聚合酶、ATP、硫酸化酶、荧光素酶、三磷酸腺苷双磷酸酶(apyrase)、腺苷5'磷酰硫酸和荧光素的存在下进行温育。个别地添加dNTP,掺入产生光信号,测量光信号。光信号指示由于成功的扩增、杂交和单碱基或多碱基延伸导致的转基因插入物/侧翼序列的存在。(当特定的基因已知时,这一技术用于触发测序,而不用于检测该基因)。
分子信标已被用于在检测中使用。简要地说,设计FRET寡核苷酸探针,其与侧翼基因组和插入DNA的接点重叠。FRET探针的独特结构导致它含有的二级结构可保持荧光和淬灭部分紧密接近。FRET探针和PCR引物(一个引物在插入DNA序列中,一个在侧翼基因组序列中)在热稳定性聚合酶和dNTP的存在进行循环扩增。在成功的PCR扩增之后,FRET探针与靶序列的杂交导致探针二级结构除去,并且荧光和淬灭部分在空间上分离。荧光信号指示由于成功的扩增和杂交产生的侧翼基因组/转基因插入序列的存在。
水解探针测定,又称(Life Technologies,Foster City,Calif.),是一种检测和定量DNA序列之存在的方法。简而言之,设计FRET寡核苷酸探针,其中一个寡聚物位于转基因内部,一个位于侧翼基因组序列中,用于事件特异性检测。FRET探针和PCR引物(一个引物位于插入DNA序列中,一个位于侧翼基因组序列中)在热稳定性聚合酶和dNTP的存在下进行循环扩增。FRET探针的杂交导致FRET探针上荧光部分与淬灭部分切割并释放。荧光信号指示由于成功的扩增和杂交产生的侧翼/转基因插入序列的存在。
KASPar测定是一种检测并定量DNA序列之存在的方法。简而言之,使用一种基于聚合酶链式反应(PCR)的测定,名为测定系统,来筛选包含靶基因座的基因组DNA样品。在本主题公开的实施中使用的测定可以使用 PCR测定混合物,该混合物包含多种引物。在PCR测定混合物中使用的引物可以包括至少一种正向引物和至少一种反向引物。正向引物含有对应于供体DNA多核苷酸的特定区域的序列,而反向引物含有对应于基因组序列的特定区域的序列。此外,在PCR测定混合物中使用的引物可以包括至少一种正向引物和至少一种反向引物。例如, PCR测定混合物可以使用对应于两个不同等位基因的两种正向引物以及一种反向引物。其中一种正向引物含有对应于内源性基因组序列的特定区域的序列。第二种正向引物含有对应于供体DNA多核苷酸的特定区域的序列。反向引物含有对应于基因组序列的特定区域的序列。这样的用于检测扩增反应的测定法是本公开的实施方案。
在一些实施方案中,荧光信号或荧光染料选自下组:HEX荧光染料,FAM荧光染料,JOE荧光染料,TET荧光染料,Cy 3荧光染料,Cy 3.5荧光染料,Cy 5荧光染料,Cy 5.5荧光染料,Cy 7荧光染料和ROX荧光染料。
在其他实施方案中,扩增反应使用合适的第二荧光DNA染料进行,其能够在流式细胞术可检测的浓度范围下对细胞DNA进行染色,并具有可以被实时热循环仪检测到的荧光发射光谱。本领域的普通技术人员应当理解,其它核酸染料是已知的并且正在被不断鉴定出来。可以使用任何合适的具有适当的激发和发射光谱的核酸染料,例如SYTOX SYBR Green 和在一个实施方案中,第二荧光DNA染料是使用浓度小于10μM,小于4μM,或小于2.7μM。
在下面的实施例中被进一步举例说明本主题公开的实施方案。但是应当理解,这些实施例仅是以举例说明的方式给出的。从上述实施方案和下面的实施例,本领域技术人员可以能够确定本公开的本质特征,并且在不脱离其精神和范围的前提下,能够对本公开的实施方案进行各种变化和修改,以使其适用于各种用途和条件。因此,除了本文显示和描述的那些实施方案之外,本领域的技术人员通过前面的描述可以显见本公开实施方案的各种修改。这些修改也意图涵盖在附加权利要求的范围之内。下文是以举例说明的方式提供的,而并不意图对本发明的范围构成限制。
实施例
实施例1:结合玉米基因组座位的锌指的设计
设计如前所述地设计锌指蛋白,来针对可靶向的玉米基因座位中已鉴定的DNA序列。参见例如Urnov et al.,(2005)Nature 435:646-551。示例性靶序列和识别螺旋如表1(识别螺旋区设计)和表2(靶位点)所示。在表2中,与ZFP识别螺旋接触的靶位点的核苷酸用大写字母表示,不接触的核苷酸用小写字母表示。为玉米基因组中全部72个选定的基因组座位均设计了锌指核酸酶(ZFN)靶位点。开发了大量的ZFP设计,并在酵母替代系统中利用在玉米中鉴定和选出的72个不同的代表性基因组靶位点对它们进行了测试,以鉴定以最高水平的效率结合的锌指。利用与锌指识别序列以最高水平效率结合的特异性ZFP识别螺旋(表1)将供体序列靶定并整合在玉米基因组中。
表1.用于玉米选定基因组座位的锌指设计(N/A表示“不可用”).
表2.玉米选定基因组座位的靶位点
将玉米代表性的基因组座位锌指设计纳入锌指表达载体中,该载体编码包含至少一个具有CCHC结构的指的蛋白。参见,美国专利公开2008/0182332。特别地,每个蛋白质的最后一个指具有用于识别螺旋的CCHC骨架。非规范锌指编码序列介由一个四氨基酸接头和来自玉米的opaque-2核定位信号与IIS型限制性内切酶FokI的核酸酶域(Wah et al.,(1998)Proc.Natl.Acad.Sci.USA95:10564-10569中序列的氨基酸384-579)融合,从而形成锌指核酸酶(ZFN)。参见美国专利7,888,121。选择具有各种功能结构域的锌指用于体内使用。设计、生产了许多ZFN并测试了它们与推定基因靶向位点的结合,它们之中如上面表2中所述的ZFN被鉴定具有体内活性,并能够在植物体中高效地结合并切割独特的玉米基因组多核苷酸靶位点。
ZFN构建体组装
使用本领域公知的技能和技术设计和完成了含有如前所述鉴定的ZFN基因表达构建体的质粒载体(参见例如Ausubel或Maniatis)。将每个ZFN编码序列融合到编码opaque-2核定位信号的序列(Maddaloni et al.,(1989)Nuc.Acids Res.17:7532),其位于锌指核酸酶的上游。将非典型的锌指编码序列融合到IIS型限制性内切酶FokI的核酸酶结构域中(Wah et al.,(1998)Proc.Natl.Acad.Sci.USA 95:10564-10569中序列的氨基酸384-579)。融合蛋白的表达由来自玉米泛素基因的强组成型启动子所驱动,其包括5'非翻译区(UTR)(Toki et al.,(1992)Plant Physiology 100;1503-07)。该表达盒还包括来自玉米过氧化物酶5基因(Per5)的3'UTR(其包含转录终止子和聚腺苷酸化位点)(美国专利公开2004/0158887)。在被克隆到构建体内的两个锌指核酸酶融合蛋白之间添加编码来自Thosea asigna病毒的核苷酸序列的自水解2A(Szymczak et al.,(2004)Nat Biotechnol.22:760-760)。
使用IN-FUSIONTM Advantage Technology(Clontech,Mountain View,CA)组装质粒载体。限制性内切核酸酶购自New England Biolabs(Ipswich,MA),并使用T4DNA连接酶(Invitrogen,Carlsbad,CA)进行DNA连接。质粒制备使用质粒试剂盒(Macherey-Nagel Inc.,Bethlehem,PA)或质粒Midi试剂盒(Qiagen)按照供应商的使用说明进行。DNA片段在用琼脂糖Tris醋酸凝胶电泳之后使用QIAQUICK GEL EXTRACTION KITTM(Qiagen)进行分离。所有连接反应的菌落最先通过小量制备DNA的限制性消化进行筛选。选出的克隆的质粒DNA通过商业测序供应商(Eurofins MWG Operon,Huntsville,AL)进行测序。序列数据的组装和分析使用SEQUENCHERTM软件(Gene Codes Corp.,Ann Arbor,MI)进行。
通用供体构建体组装
为了支持对大量靶基因座的快速测试,设计并构建了新型、灵活的通用供体系统序列。该通用供体多核苷酸序列与高通量载体构建方法和分析相兼容。该通用供体系统包括至少三个模块域:非可变的ZFN结合域,分析和用户定义特征域,和用于载体放大的简单质粒骨架。非可变的通用供体多核苷酸序列对于所有供体都是共同的,其允许设计一个可以用于所有的玉米靶位点的有限的测定集合,从而提供靶向评估的一致性并缩短分析周期。这些结构域的模块化性质允许高通量供体组装。此外,通用供体多核苷酸序列还具有其他独特特征,这些特征旨在简化下游分析和强化对结果的解释。它含有一段不对称的限制性位点序列,可以用来将PCR产物消化成诊断性预测的大小。除去包含预期在PCR扩增中会成问题的二级结构的序列。通用供体多核苷酸序列的尺寸较小(小于3.0kb)。最后,通用供体多核苷酸序列是高拷贝的pUC19骨架基础上构建的,使得及时积累大量测试DNA成为可能。
作为一个实施方案,一种包含通用多核苷酸供体盒序列的示例性质粒示于SEQ ID NO:132和图1。在另一个实施方案中,多核苷酸供体盒序列示于:pDAB111846,SEQ ID NO:133,图2;pDAB117415,SEQ ID NO:134,图3;pDAB117416,SEQ ID NO:135,图4;pDAB117417,SEQ ID NO:136,图5;pDAB117419,SEQ ID NO:137,图6;pDAB117434SEQ ID NO:138,图7;pDAB117418,SEQ ID NO:139,图8;pDAB117420,SEQ ID NO:140,图9;和pDAB117421,SEQ ID NO:141,图10。在另一实施方案中,可以构建其他的序列,它们包含具有功能性表达的编码序列或非功能性(无启动子)表达的编码序列的通用供体多核苷酸序列。表3中为如上所述的构建体注释了构成通用供体系统的各个域(非可变的ZFN结合域、分析和用户定义特征域、及简单质粒骨架)。
表3.通用供体系统载体的注释,以标识出非可变的ZFN结合域、分析和用户定义特征域、及质粒骨架
在另一个实施方案中,通用供体多核苷酸序列是一种2-3Kb的小型模块性供体系统,作为质粒递送。这是一种最小供体,包含:任意数目的ZFN结合位点;一个100-150bp的短模板区域(SEQ ID NO:142和SEQ ID NO:143),称为“DNA X”或“UZI序列”或“分析域”,其携带限制性位点和供引物设计用的DNA序列(引物被设计为Tm值比任何算得的二级结构的Tm值高大于10℃)或编码序列,和简单质粒骨架(图11)。在一个实施方案中,分析域被设计为:含有的鸟嘌呤和胞嘧啶碱基对的百分比为40-60%;不包含超过9Bp的重复序列(例如,5'-gtatttcatgtatttcat-3');不包含大于9Bp的一系列相同的碱基对;和没有二级结构,其中如Markham,N.R.&Zuker,M.(2008)UNAFold:software for nucleic acid folding and hybridization.收载于Keith,J.M编辑,Bioinformatics,第II卷.Structure,Function and Applications,number 453,收编于Methods in Molecular Biology,第1章,3–31页.Humana Press,Totowa,NJ.ISBN 978-1-60327-428-9所述地计算,二级结构的自由能小于–18kcal/mol。参见表4。在合适的ZFN结合位点处DNA双链断裂之后通过NHEJ将整个质粒插入;ZFN结合位点可以串联地并入。这种通用供体多核苷酸序列的实施方案最适合于靶位点和ZFN的快速筛选,并且供体中难以扩增的序列被最少化。
表4.对分析域组成分析下述各项:ΔG自由能、9Bp相同碱基对段(run)的数目,超过9Bp的重复序列的数目,和鸟嘌呤/胞嘧啶百分比
在进一步的实施方案中,通用供体多核苷酸序列由至少四个模块组成,包括部分ZFN结合位点、同源臂、具有大约100bp的分析片或编码序列的DNA X。这个通用供体多核苷酸序列的实施方案适用于询问多个多核苷酸靶位点处由NHEJ介导的基因插入,使用数种ZFN(图12)。
通用供体多核苷酸序列可用于所有带有明确的DNA结合域的靶向分子,有两种靶向供体插入模式(NHEJ/HDR)。这样,当通用供体多核苷酸序列与合适的ZFN表达构建体一起被递送时,供体载体和玉米基因组均会在一个由具体ZFN结合所指示的特定位置处被切割。供体一旦被线性化,就可以通过NHEJ或HDR被整合到基因组中。然后可以对载体设计进行不同的分析考量,以确定能够使靶向整合的有效递送最大化的锌指。(图13)。
实施例2:玉米转化流程
在被递送到玉米栽培品种Hi-II原生质体之前,使用PURE YIELD PLASMID MAXIPREP (Promega Corporation,Madison,WI)或PLASMID MAXI (Qiagen,Valencia,CA)按照供应商的说明从大肠杆菌培养物中为每个ZFN构建体制备质粒DNA。
原生质体分离
玉米栽培品种Hi-II悬浮细胞以3.5天进度表进行保持,收集4mL细胞压积体积(packed cell volume)(PCV)的细胞并转移到含有20mL酶溶液(0.6%PECTOLYASETM,6%CELLULASETM(“Onozuka”R10;Yakult Pharmaceuticals,Japan),4mM MES(pH 5.7),0.6M甘露醇,15mM MgCl2)的50mL无菌锥形管中(Fisher Scientific)。将培养物加盖,并用PARAFILMTM包裹并放置在平台摇床上(Thermo Scientific,可变混合平台摇臂),设置速度为10,在室温下温育16-18小时,直至原生质体释放。温育后,用显微镜评价细胞的消化质量。将消化后的细胞通过一100μm细胞滤网过滤,用10mL W5培养基[2mM MES(pH5.7),205mM NaCl,167mM CaCl2,6.7mM KCl]漂洗,然后再通过70μm和40μm的细胞滤网过滤。该100μm和40μm的细胞滤网用10mL W5培养基漂洗。将过滤的原生质体与漂洗培养基一起收集到一个50ml离心管中,终体积为大约40mL。然后向原生质体/酶溶液的底部缓慢加入8mL“重梯度溶液”[500mM蔗糖,1mM CaCl2,5mM MES(pH6.0)],使用带悬挂吊篮式转子的离心机以300-350×g离心15分钟。离心后,移出大约7-8mL原生质体带,用25mL W5清洗,并在180-200×g下离心15分钟。然后将原生质体重新悬浮在10mL MMG溶液中[4mM MES(pH 5.7),0.6M甘露醇,15mM MgCl2]。使用血球计数或流式细胞仪对原生质体进行计数,并用MMG稀释至167万个/毫升。
使用PEG转化玉米栽培种Hi-II悬浮培养递送的原生质体
将大约50万个原生质体(在300μL MMG溶液中)转移到2mL管中,与40μL DNA混合,并在室温下温育5-10分钟。接着,添加300μL新鲜制备的PEG溶液(36%PEG 4000,0.3M甘露醇,0.4M CaCl2),将混合物在室温下温育15-20分钟,定期颠倒混合。温育之后,缓慢加入1mL W5清洗液,轻柔混合细胞,通过180-200×g离心15分钟沉淀原生质体。将离心沉淀物重悬在1ml WI培养基[4mM MES(pH 5.7),0.6M甘露醇,20M KCL]中,用铝箔包裹管子并在室温下过夜温育大约16小时。
ZFN和供体转化
对每个选定的基因座,用yfp基因表达对照、仅ZFN、仅供体、以及1:10比例(重量比)的ZFN和供体混合物转染玉米原生质体。转染50万个原生质体的DNA总量为80μg。所有处理均进行了三次或六次重复。所用的YFP基因表达对照是pDAB8393(图14),其含有玉米泛素1启动子–黄色荧光蛋白编码序列–玉米Per5 3'UTR和水稻Actin1启动子–pat编码序列–玉米脂肪酶3'UTR基因表达盒。在典型的靶向实验中,将单独的4μg ZFN、或者4μg ZFN与36μg供体共转染,向每个处理加入40μg YFP报告基因构建体。通过加入一致量的YFP基因表达质粒作为补充物,可以评估多个基因座和重复处理中的转染质量。另外,通过使用一致量的YFP基因表达质粒,在对供体插入进行快速靶向分析时出现的任何技术问题可以得到快速地解决。
实施例3:通过锌指核酸酶切割玉米中的基因组座位
在转染24小时后,通过在2mL EPPENDORFTM管中以1600rpm离心收集ZFN转染的玉米栽培种Hi-II原生质体,并除去上清液。使用QIAGEN植物DNA提取试剂盒TM(Qiagen,Valencia,CA)从原生质体沉淀中提取基因组DNA。将分离的DNA重悬浮在50μL水中,并通过(Invitrogen,Grand Island,NY)确定浓度。通过0.8%琼脂糖凝胶电泳估算跑胶样品中DNA的完整性。所有样品均归一化(20-25ng/μL),以便PCR扩增产生用于测序的扩增子(Illumina,Inc.,SanDiego,CA)。设计条形码引物,用来从处理样品和对照样品扩增包含来自每个测试ZFN识别序列的区域,所述引物从IDT(Coralville,IA,HPLC纯化)购买。在23.5μL反应中使用0.2μM合适的条形码引物、ACCUPRIME PFX SUPERMIXTM(Invitrogen,Carlsbad,CA)、和100ng的基因组DNA模板,通过梯度PCR确定最佳扩增条件。循环参数为::在95℃最初预变性95℃(5min),接着是35个循环的变性(95℃,15sec)、退火(55-72℃,30sec)、延伸(68℃,1min),和最终的延伸(68℃,7min)。扩增产物在用3.5%TAE琼脂糖凝胶上进行分析,确定每个引物组合的合适退火温度,并利用合适的退火温度从对照和ZFN处理样品扩增出扩增子,如上文所述。将所有扩增子在3.5%琼脂糖凝胶上纯化,用水洗脱,并通过NANODROPTM确定浓度。为了进行二代测序,合并大约100ng来自ZFN处理的和相应玉米原生质体对照的PCR扩增子,并使用Illumina Next Generation Sequencing(NGS)进行测序。
测定了合适的ZFN在每个玉米选定基因组位点的切割活性。从处理组和对照组原生质体的基因组DNA扩增出包含ZFN切割位点的短扩增子,并对这些扩增子进行Illumina NGS。细胞NHEJ修复途径通过在切割位点处插入或缺失核苷酸(Indel)而消除ZFN诱导的切割或DNA双链断裂,因此切割位点处Indel的存在是ZFN活性的一种度量,通过NGS加以确定。使用NGS分析软件(专利公开2012-0173,153,DNA序列的数据分析)(图15)评估每一百万个高品质序列中带有Indel的序列的数目,作为靶特异性ZFN的切割活性。对于玉米选定基因组座位靶点,观察到了比对照高5-100倍的活性,这进一步被序列比对所证实,序列比对显示了每个ZFN切割位点处Indel的多样性足迹。这个数据表明,玉米选定基因组座位容易被ZFN切割。各个靶点的活性差异反映了其染色质的状态和易切割性,以及每种ZFN的表达效率。
实施例4:通过锌指核酸酶快速靶向分析多核苷酸供体序列在基因组座位内的整合
使用基于原生质体的半高通量快速靶向分析方法,验证通用供体多核苷酸序列通过非同源末端连接(NHEJ)介导的供体插入在选定的玉米靶基因组座位靶点内的靶定。对于每个玉米选定靶基因组座位,测试了3-6种ZFN设计,并通过二代测序方法测量ZFN介导的切割(图15)和通过接点内-外PCR测量供体插入(图16),对靶向进行评估。将在两个测定中均为阳性的玉米选定基因组座位鉴定为可靶向的座位。
ZFN供体插入快速靶向分析
为了确定玉米选定基因组座位靶点是否能够被靶向供体插入,将ZFN构建体和通用供体多核苷酸构建体共递送到玉米原生质体中,温育24个小时,之后提取基因组DNA进行分析。如果表达的ZFN在玉米选定基因组座位靶点和供体中均能够切割靶结合位点,线性化的供体就可以通过非同源末端连接(NHEJ)途径插入到玉米基因组的切割靶位点中。根据“内-外”PCR策略来确认在玉米选定基因组座位靶点处的靶向插入,其中“外”引物识别原有基因组座位处的序列,“内”引物则结合供体DNA内的序列。引物的设计方式使得只有当供体DNA插入在玉米选定基因组座位靶点处时,PCR测定才会产生具有预期大小的扩增产物。在插入接点的5'-和3'-端均进行内-外PCR测定。用于分析被整合多核苷酸供体序列的引物提供于表5中。
使用巢式“内-外”PCR将ZFN供体插入在靶基因座处
所有PCR扩增均使用TAKARA EX TAQ HSTM试剂盒(Clonetech,Mountain View,CA)进行。第一轮内-外PCR在20μL最终反应体积中进行,其含有1X TAKARA EX TAQ HSTM缓冲液,0.2mM dNTP,0.2μM“外”引物(表5),0.05μM“内”引物(从上述的通用供体盒设计而得),0.75单位TAKARA EX TAQ HSTM聚合酶和10ng提取的玉米原生质体DNA。然后使用如下PCR程序进行反应:94℃2min,20个循环的98℃12sec和68℃2min,随后是72℃10min,并在4℃保持。最终的PCR产物在琼脂糖凝胶上与1KB PLUS DNA LADDERTM(Life Technologies,Grand Island,NY)一起跑胶,以便可视化观察。
巢式内-外PCR在20μL最终反应体积中进行,其中含有1X TaKaRa Ex TAQ HSTM缓冲液,0.2mM dNTP,0.2μM“外”引物(表5),0.1μM“内”引物(从上述的通用供体盒设计而得,表6),0.75单位的TaKaRa Ex TAQ HSTM聚合酶,和1μL第一轮PCR产物。使用如下PCR程序进行反应:94℃2min,31个循环的98℃12sec,66℃30sec和68℃45sec,随后是72℃10min,并在4℃保持。最终的PCR产物与1KB PLUS DNA LADDERTM(Life Technologies,Grand Island,NY)一起跑胶,以便可视化观察。
表5.用于最佳基因组座位巢式内-外PCR分析的所有“外”引物的列表
表6.用于最佳基因组座位巢式内-外PCR分析的所有“内”引物的列表
表7.用于ZFN切割活性的引物
在原生质体靶向系统中开展内-外PCR测定特别具有挑战性,因为要使用大量的质粒DNA进行转染,并且大量质粒DNA保留在原生质体靶向系统中,并随后与细胞基因组DNA一起被提取。残余的质粒DNA可能稀释基因组DNA的相对浓度,降低检测的总体灵敏度,并且还可能是非特异性的异常PCR反应的重要原因。ZFN诱导的基于NHEJ的供体插入通常沿着正向或反向发生。对正向插入的DNA内-外PCR分析经常显示假阳性条带,这可能是由于靶和供体的ZFN结合位点周围具有共享的同源区域,这会导致在扩增过程中引发和延伸未整合的供体DNA。在探查反向插入产物的分析中没有见到假阳性,因此在快速靶向分析中进行的所有靶向供体整合分析均查询反向供体插入。为了进一步增加特异性并降低背景,还采用了一种巢式PCR策略。该巢式PCR策略使用第二PCR扩增反应来扩增第一PCR反应的第一扩增产物内的一个较短区域。通过使用不对称量的“内”引物和“外”引物,进一步优化了接点PCR以用于选定基因组座位处的快速靶向分析。
内-外PCR分析结果在琼脂糖凝胶上可视化。对所有的玉米选定基因组座位,“ZFN+供体处理”在5'和3'端产生了接近预期大小的条带。ZFN对照或仅供体处理在PCR中是阴性的,提示该方法特异性地评估靶位点处的供体整合。所有处理均重复3-6次,利用多个重复(在两端均≥2个)中预期的PCR产物之存在来证实靶向。供体通过NHEJ的插入常常产生强度较低的副产物,其产生是由于靶标和/或供体ZFN位点处线性化末端的加工所导致。此外,观察到不同的ZFN产生靶向整合的效率水平不同,其中某些ZFN可产生一致高水平的供体整合,而某些ZFN产生的供体整合的一致性水平较低,而其他ZFN则不会产生整合。总而言之,对于每个被测试的玉米选定基因组座位靶点,有一种或多种ZFN显示了可在玉米的代表性基因组座位靶点内实现靶向整合,这证实了这些基因座中的每一个均是可以靶定的。而且,每一个玉米选定基因组座位靶点均适于进行精确的基因转化。这些玉米选定基因组座位靶点的验证经过多次重复实验进行验证,每次重复得到了相似的结果,从而确认了上述验证过程,包括质粒设计和构建、原生质体转化、样品处理和样本分析,是可重复的。
结论
将供体质粒和一个被设计为特异性切割玉米选定基因组座位靶点的ZFN转染到玉米栽培种Hi-II原生质体中,并在24小时后收集细胞。从对照组、ZFN处理组、和ZFN与供体处理组的原生质体分离基因组DNA,通过内-外接点PCR对原生质体进行分析,显示由于ZFN切割基因组DNA,通用供体多核苷酸靶向插入(表8)。这些研究表明,该通用供体多核苷酸系统可以用来评估内源位点处的靶向和用来筛选候选的ZFN。最后,基于原生质体的快速靶向分析和新的通用供体多核苷酸序列系统提供了一种改进的系统,用来筛选基因组靶点和ZFN,以供在植物中进行精确的基因组工程化工作使用。该方法可以推广到在任何感兴趣的系统中使用任何可导入DNA双链或单链断裂的核酸酶来评估位点特异性切割和供体插入。
表8.通用供体多核苷酸序列整合在玉米选定基因组座位靶点内的结果
实施例5:玉米原生质体产生和转染
通过用细胞壁消化酶(纤维素酶,“Onozuka”R10–Yakult Pharmaceuticals,Japan;和果胶酶,320952–MP Biomedicals,Santa Ana,CA)温育玉米栽培种Hi-II悬浮细胞并用蔗糖梯度纯化,获得原生质体。为了进行转染,原生质体用MMG(MES pH6.0,0.6M甘露糖醇,15mM MgCl 2)稀释到浓度为167万/ml,并将等份的300μL原生质体(~500,000)加到无菌的2ml管中,向每个2ml管添加总浓度为40μg的质粒DNA(包含ZFN/多核苷酸供体转基因组合表达盒的YFP转基因表达盒、ZFN转基因表达盒、多核苷酸供体转基因表达盒),轻轻混合并在室温下温育5-10分钟。接着,向原生质体/DNA溶液添加300μL PEG 4000,将混合物颠倒混合,直至PEG 4000与原生质体/DNA溶液完全混匀。接着,将原生质体/DNA/PEG混合物在室温下温育15-20分钟。温育之后,用1ml W5(2mM MES pH6.0,205mMNaCl,167mM CaCl2,6.7mM KCl)清洗原生质体/DNA/PEG混合物,并在180-200×g离心15分钟。去除上清液后,加入1ml WI培养基(4mM MES pH6.0,0.6M mannitol,20mMKCl)重悬细胞原生质体离心沉淀。重悬后的离心沉淀用铝箔覆盖,温育过夜。使用Beckman-Coulter公司(Brea,CA)的Quanta FlowcytometerTM计算原生质体转染效率,算出的转染效率在10-50%的范围内。所有转染处理均进行六次重复。
玉米栽培种B104原生质体的分离采用与前述玉米栽培种Hi-II来源的原生质体相似的转染流程。如下所述从幼嫩苞皮组织获得原生质体:将苞皮人工切成薄条(大约0.5mm),然后横切成薄片。将切片组织转移至装有25mL酶溶液的无菌锥形瓶中,并将其在干燥箱中放置15分钟。然后将烧瓶加盖,用铝箔包裹,并在室温下在定轨摇床上以最低速度震荡过夜。
实施例6:整合在工程化着陆平台(Engineered Landing Pad)基因组位点内的供体多核苷酸的快速靶向分析
供体插入在玉米的工程化基因座:用一种分析来证实一5Kb供体插入在如美国专利申请No.2011/0191899所述的工程化着陆平台1(ELP1)基因组靶点内。供体DNA通过NHEJ整合方法被插入到玉米栽培种Hi-II品系原生质体的基因组中(该品系,“106685[1]-007”,是通过pDAB106685的转化和整合而产生的)。供体整合在ZFN1和ZFN3锌指结合位点内(图18)。用于NHEJ介导整合在ELP1基因组靶点内的方法要求ELP1靶点和供体质粒含有相同的ZFN位点(ZFN1或ZFN3)。当供体多核苷酸序列和ZFN被转染进原生质体细胞内时,ZFN切割ELP1基因组靶点和质粒供体DNA,从而产生相同的末端。所得的相同末端通过NHEJ介导的细胞修复作用被连接,结果质粒供体DNA靶向插入在ELP1基因组靶点内。使用两种不同的ZFN-供体摩尔比(1:1和1:10)均证实了ELP1基因组靶点的靶定。使用基因座破坏测定(locus disruption assay)和内-外PCR确认了供体整合的结果,但不对称PCR引物浓度不包括在内。供体多核苷酸序列的插入可以在两个方向上发生,并且该内-外PCR被设计为这两个方向都检测。
破坏测定:破坏测定是一种水解探针测定(与TaqmanTM相似),其测量基因座DNA序列ZFN结合位点是否已被修改或重排。相应地,测定ZFN结合位点的完整性。ZFN介导的供体插入或切割,随后是NHEJ修复,导致ZFN结合位点的丧失及可检测的qPCR信号降低(参见,美国专利公开2014/0173783,其通过引用并入本文)。图19提供了在ZFN1和ZFN3位点处切割ELP1、以及供体序列靶向整合在这些位点内的结果。通过递送ZFN多核苷酸测定ZFN1位点,相关的ZFN与供体多核苷酸的比例为1:1和1:10。结果表明ELP1的ZFN1位点被破坏,从而提示该位点可能被靶向。同样地,ELP1的ZFN3位点被破坏,也提示该位点可能被靶向。此实验的所有处理均进行6次重复,数据作为平均值结果呈现。
内-外PCR测定:为了确认ELP1的ZFN1和ZFN3处靶向供体的插入,对从对照原生质体样品(例如,仅用ZFN多核苷酸或供体多核苷酸处理的样品)和用ZFN和供体多核苷酸二者处理的原生质体样品分离基因组DNA进行内-外PCR。PCR引物被设计为沿着任一方向扩增和检测供体的插入。内-外PCR结果表明,对于所有被试样品(例如1:1和1:10比例的供体:ZFN),ELP1的ZFN1和ZFN3位点处均有靶向供体插入。ZFN1位点在6次中有4次被靶向,而ZFN3位点在6次中有3次被靶向。正向和反向的供体插入均通过内-外PCR测定被检测到。对PCR材料的测序显示了预期的靶标-供体接点序列,以及供体和/或靶标在连接之前被加工的接点(图20)。
实施例7:供体多核苷酸在内源玉米基因座内的整合的快速分析
玉米事件DAS-59132(本文称作E32)的基因组座位被靶向用于多核苷酸供体插入,该事件如美国专利公开2014/0173783所述,其通过引用并入本文。使用E32ZFN6(pDAB105906)将含有add-1转基因的5Kb的供体多核苷酸(pDAB100651)靶向到玉米的内源基因座(E32)处。利用快速靶向分析,通过对插入了供体的多核苷酸的5’和3’端进行新的内-外PCR测定(图21),确认供体多核苷酸在玉米基因组内的位点特异性整合。
应用快速靶向分析作为原生质体转染系统的内-外PCR测定是特别有挑战性的,因为原生质体转染系统是一个瞬时转化过程。因此,被递送到原生质体细胞的大幅过量的质粒DNA会保留在系统中,并可以和细胞基因组DNA一起被提取。递送大量的质粒DNA不仅会稀释基因组DNA的有效浓度,从而使基因组靶向的检测变得困难,还会导致非特异性的PCR反应,产生假阳性。
在快速靶向分析内-外PCR测定的开发过程中,鉴定出了原生质体系统中假阳性的一个主要来源。正如这些研究过程中所证明的,基于NHEJ的供体插入能够沿着两个不同的方向发生,供体能够沿着正向或反向插入到基因组中。内-外PCR扩增和正向插入的分析常常造成假阳性的强而深的扩增子。相反,内-外PCR扩增和反向插入的分析未产生大量的假阳性扩增子。应当指出的是,供体多核苷酸和内源E32基因座共享相同的ZFN结合位点,这可能导致PCR交叉反应(如图22所示)。假阳性很可能是由于交叉反应产生的副产物,该交叉反应是由于模板的复制、产生组入有ZFN结合位点的延伸扩增链所导致的。所得的扩增链随后可能在下一个PCR循环中结合内源基因组序列的ZFN结合位点,或者结合多核苷酸供体序列,导致一被PCR反应扩增的假阳性模板。
不对称巢式内-外(ANIO)PCR:为了进一步减少非特异性PCR扩增,设计了巢式内-外PCR策略,使其可以利用第二轮内-外PCR扩增来扩增第一轮内-外PCR扩增子内的一个区域。后续的PCR扩增进一步提高了基因组座位内供体靶向和整合的特异性和检测。在设计和实现巢式PCR反应的过程中,鉴定了另一个用于降低非特异性扩增的新改进。由于存在大量供体质粒DNA,怀疑可结合供体DNA的“内”引物可能对假阳性具有重要贡献。通过降低“内”引物与“外”引物浓度相比的浓度,假阳性显著降低。最终的不对称巢式内-外(ANIO)PCR被用来证明供体多核苷酸在原生质体中的玉米E32基因座处的靶向。所有PCR引物均基于一种阳性对照质粒来设计,该质粒是为了模拟E32基因座处的靶向插入(表9)而构建的。
表9.下表显示了用于ANIO PCR的引物列表
具体地讲,第一轮内-外PCR在20μL最终反应体积中进行,其含有1x TaKaRa Ex Taq HS缓冲液TM,0.2mM dNTP,0.2μM“外”引物,0.05μM“内”引物,0.75单位的TaKaRa Ex Taq HSTM聚合酶,和10ng提取的玉米原生质体DNA。PCR反应采用一PCR程序来完成,该程序为:94℃2min,20个循环的98℃12sec和68℃2min,随后是72℃10min,并保持在4℃。
巢式(或第二轮)内-外PCR反应在20μL的最终反应体积中进行,其包含1x TaKaRa Ex Taq HS缓冲液TM,0.2mM dNTP,0.2μM“外”引物,0.1μM“内”引物,0.75单位TaKaRa Ex Taq HS聚合酶TM和1μL第一轮PCR产物。PCR反应采用94℃2min,31个循环的98℃12sec,66℃30sec和68℃45sec,随后是72℃10min,并保持在4℃来完成。最终的PCR产物与1Kb Plus DNA LadderTM(Life Technologies,Grand Island,NY)一起在琼脂糖凝胶上跑胶以便可视化。
快速靶向分析检测到供体多核苷酸沿着反向插入在E32基因组靶座位内。在所有用ZFN和供体组合处理的样品中,6次反应中的6次均对5'和3'端产生了具有预期大小的扩增子(相比之下,对照组没有得到供体多核苷酸序列的位点特异性整合)。对于3'端的扩增,在琼脂糖凝胶上观察到了少量拖尾或阶梯现象。这一观察结果可能是由于对NHEJ修复之前产生的DNA断裂末端的加工所导致的。此外,由在含有用于3’端扩增的“仅供体”的对照样品中,观察到了非特异性扩增子的扩增。非特异性扩增子的分子量比阳性对照的扩增子的预期大小要小。尽管如此,供体多核苷酸供体被成功地整合在E32基因组靶座位内,并且采用快速靶向分析高效地鉴定和检测了位点特异性整合。
实施例8:整合在内源大豆基因座内的供体多核苷酸的靶向分析
使用上述的转化方法将设计好的ZFN转化进大豆原生质体中。通过如在美国专利公开2014/0173783中描述的位点破坏测定,对各种ZFN切割FAD2位点的效率进行了评估。此外,使用内-外PCR测定评估了供体序列在锌指核酸酶介导下在FAD2基因座内的整合,并对所产生的PCR扩增子进行测序以表征大豆基因组内的供体整合。
实验包括下述处理组:仅有供体载体、仅有ZFN载体、或ZFN与供体载体组合(表10)。此外,实验包括阴性对照处理组,即未转化的细胞或用对照载体pDAB7221转化的细胞(图23),该质粒包括一个由CsVMV启动子驱动的绿色荧光蛋白表达盒,该表达盒两侧翼是高拷贝数质粒内的AtuORF243'-UTR。在转染后大约18-24小时收集被转染的样品。初步数据表明,F2,pDAB115601质粒中所含的ZFN,具有高活性,因此,将这种ZFN质粒在所有后续实验中用作阳性对照。
如表10所详述的,转化实验包含总共80μg DNA,并根据需要添加质粒PDB7221以使DNA的总量达到80μg。供体载体与ZFN表达质粒的比例为大约10:1。每个实验或处理包括6个独立处理和分析的重复实验。对ZFN的评价分两组实验进行。
表10.实验设计。在两组实验(F2ZFN1-3和F2ZFN4-7)中对ZFN质粒进行评价。靶向实验使用适合于ZFN质粒的供体载体。每个处理进行6个重复。
对靶向的分析:使用基因座破坏测定分析靶向实验的DNA样品,以检测FAD2ZFN切割位点处的修饰并评估NHEJ介导的靶向。设计qPCR测定,用以测量FAD2靶标内完整的ZFN结合位点。ZFN介导的供体插入或切割以及随后的NHEJ修复导致ZFN结合位点的丧失,并随后导致可检测的qPCR信号减小。与单独供体处理相比,具有显著切割活性的ZFN导致扩增子产生的信号减小。在基因座破坏测定中使用的引物和探针如表11所示,它们在FAD2基因座上的相对位置如图24所示。
在合适的供体载体的存在下,用FAD22.3ZFN2_WT ZFN(两个实验)和FAD22.6ZFN ZFN4_HF(一个实验)以及F2ZFN5_HF(两个实验)处理原生质体,导致其信号与从完整序列(仅有供体)获得的信号相比有统计学显著性的减小。
表11.用于破坏PCR(disruption PCR)的引物和探针
基因座特异性内-外PCR:为了确认靶向供体插入,所有处理组的DNA均进行基因座特异性的内-外PCR测定。实验中的供体载体设计为包含所有被测试FAD2基因座内靶向整合的ZFN的结合位点。ZFN和供体共同递送到大豆细胞内,导致靶点处和供体载体内的ZFN结合位点被切割,供体随后通过非同源末端连接机制整合到被切割的FAD2基因座内。在整合到FAD2基因座内之前,ZFN切割所生成的FAD2染色体位点及线性化供体载体的末端会受到加工,并可能导致不完美的末端连接产物。靶点处的靶向整合的确认是根据“内-外”PCR策略进行的,其中“外”引物识别天然基因组座位处的序列,而“内”引物结合供体DNA内的序列。在插入接点的5'和3'端均进行内-外PCR测定。
所有被测试的ZFN均在至少一个实验中显示了某些供体片段被靶向并整合到FAD2大豆基因座内的证据,如根据供体和ZFN样品中的PCR产物所确定的。使用如下ZFN:F2ZFN2_WT,F2ZFN2_HF和F2ZFN4_HF的供体整合靶向的结果是可重复的,因为在6次实验重复中的至少2次中在5’和3’端均产生了PCR产物(表12)。
表12.在大豆原生质体的FAD2基因座处NHEJ靶向的总结。显示了每个实验或处理在独立的靶向实验中对内-外PCR呈阳性的重复数目。
内-外PCR产物的测序:将使用pDAB1115620和F2ZFN2_WT或者pDAB1115620和F2ZFN2_HF完成的每个内-外PCR靶向实验的两个扩增子(具有期望大小)克隆到质粒中。所得质粒用桑格测序法进行测序。将序列与参考序列进行比对,在参考序列中,将预期通过FokI切割产生的各个单链4bp末端加以复制,以反映所有可能的末端组合。从所获得的23个克隆序列中发现了10个独特的序列模式(图25)。所有序列模式均保留位于ZFN结合位点之间的部分FAD2基因组参考序列(GAAATTTC),但是与FAD2基因组参考序列相比,这些序列模式还具有缺失。序列4WT1和4WT4含有延伸到GAAATTTC序列3’端的ZFN结合位点内的缺失。两个序列,1HF4和6HF4,具有单碱基插入。观察到的DNA序列模式证实,供体DNA靶向到了大豆FAD2基因座内。
虽然在某些实施方案中对本发明的各方面进行了描述,但是在本公开的精神和范围内还可以进一步修改。因此,本申请意图涵盖使用其一般原理对本发明实施方案的任何改变、使用或修改。此外,本申请意图涵盖在本实施方案所属领域的已知或通常实践中会发生的与本公开的背离,并且其也包含在附加权利要求的范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 阻断UBE2M介导Neddylation修饰在肾细胞癌治疗中的应用的制造方法与工艺
- 无载体共组装肿瘤靶向抗癌纳米药物及其制备方法与应用与流程
- 一种用于判断靶向单克隆抗体治疗肿瘤疗效的预测分子的制造方法与工艺
- 一种靶向CD47的免疫检查点抑制剂药物组合物及其制备方法与流程
- 具有淋巴靶向功能的生物自组装纳米晶注射剂及制备方法与流程
- 用于检测肝癌的基因标志物及其用途的制造方法与工艺
- 特异性shRNA筛选及其靶向Ang‑2基因抑制肺癌细胞的验证方法与流程
- 一种多功能自动化生物细胞提取分离控制系统及采用该系统实现的细胞提取分离方法与流程
- 一种融合蛋白及光敏剂复合物及其制备方法和应用与流程
- 具有可视化和显示光斑边界功能的光动力治疗系统的制造方法与工艺