一种靶向抗肿瘤药物F16与苯丁酸氮芥合成的化合物及其制备方法与流程
本发明涉及一种抗肿瘤药物化合物,尤其涉及一种靶向抗肿瘤药物F16与苯丁酸氮芥的化合物及其制备方法。
背景技术:
癌症是严重危害人类健康的一大顽症,现已成为仅次于心血管病的第二大杀手,所以寻找抗肿瘤药物和研究其作用机理,具有重要意义。
线粒体是细胞内重要的细胞器,在细胞代谢过程中扮演了重要角色,是生命体的“能量工厂”,参与了能量产生细胞凋亡、细胞癌变等多种代谢过程。线粒体结构和功能的改变,不仅会干扰细胞的生长、代谢和增殖过程,还会引起细胞凋亡。因此,以线粒体为靶点的新型抗肿瘤药物的研发,已成为一大研究热点。
F16是近年来发现的一种高度疏水的、DLC(delocalize lipid cationic)药物,F16是由乙烯基将一个吡啶环铜一个吲哚环连接而成,其结构如下所示:
F16能有效延长线粒体恢复周期,降低线粒体最大产热量和线粒体活性 恢复期速率常数,F16对于线粒体的作用机制是通过打开舌头转换孔道,破坏线粒体质子动力势,从而干扰线粒体代谢产热。
但是目前一些以线粒体为靶点治疗癌症的药物化合物,普遍存在选择性低和多耐药性的问题,为此,开发一种基于F16的衍生型抗肿瘤药物化合物即成为必要。
技术实现要素:
本发明的特征和优点在下文的描述中部分地陈述,或者可从该描述显而易见,或者可通过实践本发明而学习。
为克服现有技术的问题,本发明提供一种靶向抗肿瘤药物F16与苯丁酸氮芥合成的化合物及其制备方法,以线粒体为靶点治疗癌症,克服抗癌药物选择性低和多耐药性的问题,达到通过细胞凋亡的途径清除癌细胞的效果。
为达到上述发明目的,本发明提出以下的技术方案:
靶向抗肿瘤药物F16与苯丁酸氮芥的化合物,具有通式Ⅰ:
其中,Y为NH、O、S;A、B、C、D、E为相同或不相同的氢基、卤基、烷基、卤仿基、烷氨基、醚基或巯醚基,n=0,1~18。
本发明提供的通式Ⅰ的化合物,其烷基为-R,烷氨基为-NH-R,醚基为-O-R,巯醚基为-S-R;其中,R=CH3(CH2)m,m=0,1~10。
本发明还提供一种制备上述通式Ⅰ化合物的方法,包括以下步骤:
S1、取化合物α,
与4-甲基吡啶反应得到化合物β:
其中X为卤基,n=0,1~18
S2、化合物β经二碳酸二叔丁酯保护得到化合物γ:
S3、化合物γ经与化合物δ:
在哌啶作用下反应得到化合物ε:
其中,Y为NH、O、S;A、B、C、D、E为氢基、卤基、烷基、卤仿基、烷氨基、醚基或巯醚基
S4、化合物ε经盐酸脱保护得到化合物η:
S5、化合物η经与化合物θ:
缩合反应得到通式Ⅰ的化合物。
上述制备方法中,在步骤S1中,化合物α与4-甲基吡啶在无水醇或醚溶剂中,加热回流反应,反应完全后,冷却反应体系至室温,过滤,用上述溶剂洗涤;所述无水醇或醚溶剂是无水甲醇、无水乙醇或无水乙醚。
上述制备方法中,在步骤S2中,将化合物β、四氢呋喃、碳酸钠溶于水,室温搅拌后缓慢滴加二碳酸二叔丁酯,在25-30℃反应4小时;过滤,将滤液旋干,然后加入反应量体积的二氯甲烷与无水醇或醚进行搅拌,再过滤,将滤液旋干得到化合物γ;所述无水醇或醚溶剂是无水甲醇、无水乙醇或无水乙醚。
上述制备方法中,所述二氯甲烷与无水醇或醚的体积比为[8~15]:1。
上述制备方法中,在步骤S3中,将化合物γ、化合物δ、无水醇或醚溶剂、哌啶升温至50℃反应,反应完全后,将反应体系旋干过柱,然后加入反应量体积的二氯甲烷与无水醇或醚进行搅拌,再过滤,将滤液旋干得到化合物ε;所述无水醇或醚溶剂是无水甲醇、无水乙醇或无水乙醚。
上述制备方法中,所述二氯甲烷与无水醇或醚溶剂的体积比为[25~35]:1。
上述制备方法中,在步骤S4中,在化合物ε中添加甲醇,室温滴加浓盐酸,室温反应2~7小时,将反应体系旋干得到化合物η。
上述制备方法中,在步骤S5中,将化合物η、N,N-二甲基甲酰胺、 N,N-二异丙基乙胺在室温下搅拌,然后加入化合物θ、以及2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,在20-25℃反应,向反应体系中加入水,用二氯甲烷萃取,有机相再用水洗涤,干燥,浓缩过柱,制得通式Ⅰ化合物。
通过阅读说明书,本领域普通技术人员将更好地了解这些技术方案的特征和内容。
【附图说明】
下面通过参考附图并结合实例具体地描述本发明,本发明的优点和实现方式将会更加明显,其中附图所示内容仅用于对本发明的解释说明,而不构成对本发明的任何意义上的限制,在附图中:
图1为本发明所提供的通式Ⅰ化合物的合成路径图。
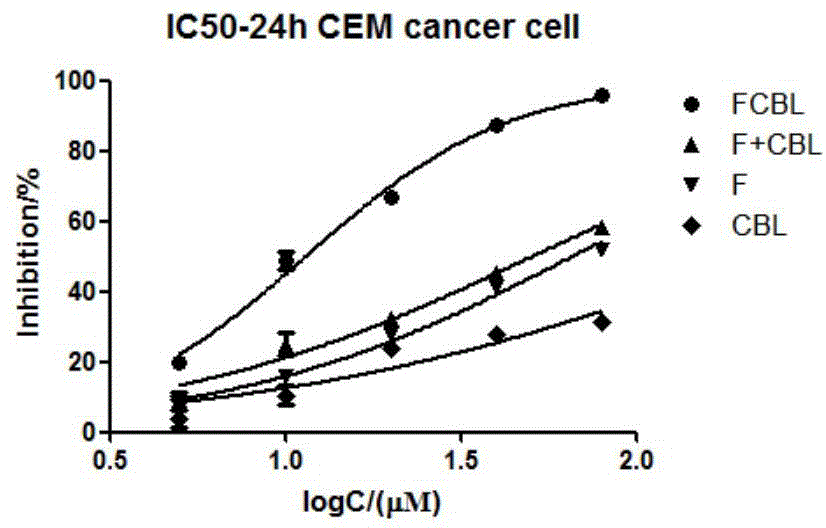
图2-1所示是本发明实施一所制得化合物对CEM细胞的抑制曲线图;
图2-2所示是本发明实施一所制得化合物对Romas细胞的抑制曲线图;
图2-3所示是本发明实施一所制得化合物对HepG2细胞的抑制曲线图;
图2-4所示是本发明实施一所制得化合物对HeLa细胞的抑制曲线图;
图2-5所示是本发明实施一所制得化合物对L-O2细胞的抑制曲线图;
图3-1所示是本发明实施一所制得化合物的线粒体定位激光共聚焦显微镜图;
图3-2所示是对比化合物的线粒体定位激光共聚焦显微镜图;
图3-3所示是另一对比化合物的线粒体定位激光共聚焦显微镜图;
图3-4所示是再一对比化合物的线粒体定位激光共聚焦显微镜图。
【具体实施方式】
本发明提供一种具有通式I的靶向抗肿瘤药物F16与苯丁酸氮芥合成 的化合物:
其中,Y为NH、O、S;A、B、C、D、E为相同或不同的氢基、卤基、烷基、卤仿基、烷氨基、醚基或巯醚基,n=0,1~18。
本发明提供的通式Ⅰ的化合物中,其烷基为-R,烷氨基为-NH-R,醚基为-O-R,巯醚基为-S-R;其中,R=CH3(CH2)m,m=0,1~10。
更具体地说,在上述通式I中:Y代表NH、O、S中的任意一种;A、B、C、D、E则代表X、R、YR、CX3、中的任意一种;其中X代表氢或卤素,即H、F、Cl、Br、I中的任意一种;R为烷基CH3(CH2)m,m=0,1~10。
请参照图1,本发明还提供一种制备上述通式Ⅰ化合物的方法,包括以下步骤:
S1、取化合物α:与4-甲基吡啶反应得到化合物β:
其中X为卤基,n=0,1~18;化合物α与4-甲基吡啶在无水醇或醚溶剂中,加热回流反应,反应完全后,冷却反应体系至室温,过滤,再用少量无水醇或醚溶剂洗涤。
S2、化合物β经二碳酸二叔丁酯保护得到化合物γ:
具体为,将化合物β、四氢呋喃、无水碳酸钠溶于水,室温搅拌后缓慢滴加二碳酸二叔丁酯,在25-30℃反应4小时;过滤,将滤液旋干,然后加入适量体积的二氯甲烷与无水醇或醚溶剂进行搅拌,再过滤,将滤液再旋干得到化合物γ;其中,二氯甲烷与无水醇或醚溶剂的体积比为[8~15]:1。
S3、化合物γ经与化合物δ:在哌啶作用下反应得到 化合物ε:
具体为,将化合物γ、化合物δ、无水甲醇、哌啶升温至50℃反应,反应完全后,将反应体系旋干过柱,然后加入适量体积的二氯甲烷与甲醇进行搅拌,再过滤,将滤液再旋干得到化合物ε;其中,氯甲烷与甲醇的体积比为[25~35]:1。
其中,Y为NH、O、S;A、B、C、D、E为相同或不相同的氢基、卤基、烷基、卤仿基、烷氨基、醚基或巯醚基。
S4、化合物ε经盐酸脱保护得到化合物η:
具体为,在化合物ε中添加甲醇,室温滴加浓盐酸,室温反应2至7小时,将反应体系旋干得到化合物η。
S5、化合物η经与化合物θ:
缩合反应,即得到通式Ⅰ的化合物。具体为,将化合物η、N,N-二甲基甲酰胺、N,N-二异丙基乙胺在室温下搅拌,然后加入化合物θ、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,在20-25℃反应,向反应体系中加入水,用二氯甲烷萃取,有机相再用水洗涤,干燥,浓缩过柱得到通式Ⅰ的化合物。
下面通过具体的实施例,进一步阐述制备通式I化合物的方法:
实施例1
首先,取化合物1(3-溴丙胺氢溴酸盐)与4-甲基吡啶反应,化合物1结构如下所示:
具体实施时,在反应瓶中分别加入4-甲基吡啶(2g),3-溴丙胺氢溴酸盐(1.0eq),无水甲醇(或无水乙醇、无水乙醚)10ml,然后加热回流反应过夜。反应完全,冷却反应体系至室温,过滤,用少量甲醇洗涤,干燥得到白色固体即化合物2(5.0g);本实施例中,化合物2的结构如下所示:
紧接着,在反应瓶中加入化合物2(2.27g)、四氢呋喃20ml、无水碳酸钠(2.0eq)溶于水20ml,室温搅拌30min,然后缓慢滴加二碳酸二叔丁酯(1.1eq)溶于四氢呋喃10ml,在25-30℃反应4小时。过滤,将滤液旋干,然后加入适量体积的二氯甲烷与甲醇(体积比8:1~15:1,优选10:1)搅拌一段时间,过滤,滤液再旋干得到浅粉色油状物即化合物3(2.31g),化合物3的结构如下所示:
随后,将化合物3与化合物4(吲哚-3-甲醛)反应,
其具体步骤为:在反应瓶中分别加入化合物3(2.31g)、吲哚-3-甲醛(1.0eq)、无水甲醇50ml,然后加入哌啶(0.9eq)、升温至50℃反应过 夜。反应完全,将反应体系旋干过柱,然后加入适量体积的二氯甲烷与甲醇(体积比25:1~35:1,优选30:1)进行搅拌,再过滤,将滤液再旋干得到深红色半固状物即化合物5(1.27g),化合物5的结构如下所示:
再接着,在反应瓶中加入化合物5(1.27g)、甲醇10ml,室温滴加浓盐酸5ml,室温反应5小时,将反应体系旋干得到深红色半固状物即化合物6(1.0g),化合物6的结构如下所示:
再后,将化合物6与化合物7(苯丁酸氮芥)反应,
其具体步骤为:在反应瓶中加入化合物6(100mg)、N,N-二甲基甲酰胺10ml、N,N-二异丙基乙胺(3.0eq),室温搅拌20min,然后加入苯丁酸氮芥(1.2eq)、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1.2eq),在20-25℃反应过夜。向反应体系中加入水20ml,用二氯甲烷20ml×3萃 取,有机相再用水20ml×2洗涤,干燥,浓缩过柱得到100mg黄色泡沫状固体即F16与苯丁酸氮芥合成的化合物8,其结构式如下:
实施例2
制备化合物3的方式与实施例1中相同,在此不再赘述。
取化合物3与化合物9(6-氟吲哚-3-甲醛)反应,
其具体步骤为:在反应瓶中分别加入化合物3(1.0g)、6-氟吲哚-3-甲醛(1.0eq)、无水甲醇30ml,然后加入哌啶(0.9eq),升温至50℃反应过夜,反应完全,将反应体系旋干过柱,然后加入适量体积的二氯甲烷与甲醇(35:1)进行搅拌,再过滤,将滤液再旋干得到深红色半固状物即化合物10(0.7g),化合物10的结构如下所示:
然后,在反应瓶中加入化合物10(0.5g)、甲醇5ml(或乙醇、乙醚),室温滴加浓盐酸2.5ml,室温反应3小时。将反应体系旋干得到深红色半固状物即化合物11(0.4g),化合物11的结构如下所示:
取化合物11与化合物7(苯丁酸氮芥)反应,其具体步骤为:在反应瓶中加入化合物11(100mg)、N,N-二甲基甲酰胺10ml、N,N-二异丙基乙胺(3.0eq),室温搅拌20min,然后加入苯丁酸氮芥(1.2eq)、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1.2eq),在20-25℃反应过夜。向反应体系中加入水20ml,用二氯甲烷20ml×3萃取,有机相再用水20ml×2洗涤,干燥,浓缩过柱得到96mg黄色泡沫状固体即F16与苯丁酸氮芥合成的化合物12,其结构式如下:
实施例3
制备化合物3的方式与实施例1中相同,在此不再赘述。
取化合物3与化合物13(5-氟吲哚-3-甲醛)反应,
其具体步骤为:在反应瓶中分别加入化合物3(1.0g)、5-氟吲哚-3-甲醛(1.0eq)、无水甲醇30ml,然后加入哌啶(0.9eq),升温至50℃反应过夜。反应完全,将反应体系旋干过柱,然后加入适量体积的二氯甲烷与甲醇(25:1)进行搅拌,再过滤,将滤液再旋干得到深红色半固状物即化合物14(0.75g),化合物14的结构如下所示:
随后,在反应瓶中加入化合物14(0.5g)、甲醇5ml,室温滴加浓盐酸2.5ml,室温反应3小时,将反应体系旋干得到深红色半固状物即化合物15(0.42g),化合物15的结构如下所示:
最后取化合物15与化合物7反应,其具体步骤为:在反应瓶中加入化合物15(100mg)、N,N-二甲基甲酰胺10ml、N,N-二异丙基乙胺(3.0eq), 室温搅拌20min,然后加入苯丁酸氮芥(1.2eq)、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1.2eq),在20-25℃反应过夜。向反应体系中加入水20ml,用二氯甲烷20ml×3萃取,有机相再用水20ml×2洗涤,干燥,浓缩过柱得到100mg黄色泡沫状固体即F16与苯丁酸氮芥合成的化合物16,其结构式如下:
实施例4
制备化合物3的方式与实施例1中相同,在此不再赘述。
取化合物3与化合物17(5-甲氧基吲哚-3-甲醛)反应,
其具体步骤为:在反应瓶中分别加入化合物3(1.0g)、5-甲氧基吲哚-3-甲醛(1.0eq)、无水甲醇30ml,然后加入哌啶(0.9eq),升温至50℃反应过夜。反应完全,将反应体系旋干过柱,然后加入适量体积的二氯甲烷与甲醇(30:1)进行搅拌,再过滤,将滤液再旋干得到深红色半固状物即化合物18(0.80g),化合物18的结构如下所示:
随后,在反应瓶中加入化合物18(0.5g)、甲醇5ml,室温滴加浓盐酸2.5ml,室温反应3小时,将反应体系旋干得到深红色半固状物即化合物19(0.45g,),化合物19的结构如下所示:
最后,取化合物19与化合物7反应,其具体步骤为:在反应瓶中加入化合物19(100mg)、N,N-二甲基甲酰胺10ml、N,N-二异丙基乙胺(3.0eq),室温搅拌20min,然后加入苯丁酸氮芥(1.2eq)、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1.2eq),在20-25℃反应过夜。向反应体系中加入水20ml,用二氯甲烷20ml×3萃取,有机相再用水20ml×2洗涤,干燥,浓缩过柱得到100mg黄色泡沫状固体即即F16与苯丁酸氮芥合成的化合物20,其结构式如下:
药物性能评价
1.体外抗肿瘤效应评价
针对前述实施例一所制得的化合物8的进行体外抗肿瘤效应评价。鉴于苯丁酸氮芥是广谱抗癌烷化剂,本实施例采用CEM、Romas、HeLa和HepG2细胞对其进行药效评价,同时LO2肝脏细胞对其进行毒性检测。
取对数生长期的细胞,根据细胞的大小接种4~40×103个于96孔板上,待生长24小时后,弃上清,然后按以下分组给药:肿瘤细胞设不加药组和加药组(浓度2.5~160μM对肿瘤细胞,浓度50~800μM对LO2细胞),F为化合物6,CBL为化合物7(苯丁酸氮芥),F+CBL为F和CBL等浓度的组合,FCBL为合成化合物8。每组设4~6个复孔,培养24或72小时,弃上清,加入100μl含0.5mg/ml的MTT(四氮唑盐)无血清培养液培养4h,加入100μl DMSO(二甲亚砜),放置于微型振荡仪上振荡10min,再置于酶标仪上570nm处检测OD值。正常人细胞系LO2做对照。每次实验均重复3次。
结果显示,随着药物浓度增加,与相应不加药对照组比较,细胞增殖活性分别下降,说明化合物呈浓度依赖性抑制肿瘤细胞细胞增殖。而对正常肝细胞系L-O2细胞的增殖活性未有变化,显示出该化合物对正常细胞具有低毒特性(如表1,图2-1至图2-5所示)。
表1 不同细胞的IC50值(24h)及不同化合物IC50比值
2.线粒体定位确证
对化合物8的线粒体进行定位确证。将Hela接板光学培养皿后,将线粒体定位试剂Mitotracker和化合物8一块与细胞进行孵育1-2小时,激光共聚焦显微镜(600X)观察拍照,红色和绿色荧光进行观察共定位;同时将对比化合物也进行相应的定位,然后同等条件下观察拍照,得到的结果如图3-1到3-4所示。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!