靶向PLCε基因RNA干扰重组慢病毒载体及其构建方法与流程
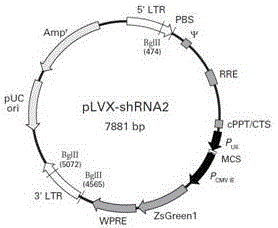
本发明涉及靶向人源磷脂酶Cε(PhospholipaseC,PLCε)基因的RNA干扰重组慢病毒载体及其构建,属于生物医药
技术领域:
。
背景技术:
:肿瘤相关基因中,Ras基因最为常见,参与细胞增殖信号传导,编码鸟嘌呤核苷酸结合蛋白(G蛋白)。Ras激活后,G蛋白只保留GTP结合活性而无水解活性,故持续激活,有丝分裂信号持续传入,细胞过度增殖。在Ras信号通路众多下游效应蛋白中,近年来磷脂酶C(phospholipaseC,PLC)家族中一个新的亚型PLCε被发现,除与其他PLC同有典型结构域X、Y和C2,其羧基端还具有特异性的Ras结合结构域和氨基端鸟苷酸交换因子结构域。PLCε在化学致癌物诱导鼠皮肤肿瘤的过程中发挥促进作用,并与肿瘤增殖有关。随后的研究表明,PLCε的促进作用可能归因于其扩大了炎症反应。PLCε通过增加炎症反应和血管形成促进鼠肠道肿瘤的发生。PLCε作为Ras原癌基因的效应子,其在肿瘤发生和进展的作用至关重要。RNA干扰技术能特异性降解mRNA,沉默靶基因,在转录后水平抑制基因的表达,从而可用来进行基因功能的研究和药物靶标的研究。目前通过siRNA诱导哺乳动物细胞内RNA干扰的操作方法有两种,分别为化学合成的siRNA和细胞内表达的siRNA。化学合成法人工制备siRNA,一般是分别合成21个碱基的正义链和反义链,体外适当的温度下使互补链配对,形成siRNA。然后,采用传统转染的方法,如Lipofectamine转染、磷酸钙、电打孔等将siRNA导入细胞,诱导RNA干扰。此方法操作简单、快速,但引起的RNA干扰效应往往是暂时的,难以持久。因此,目前更倾向于在哺乳动物细胞内表达siRNA,以维持长久的RNA干扰效应。细胞内表达法是将表达siRNA的载体转染细胞,在细胞内表达siRNA。慢病毒载体作为常用的病毒载体之一,其特点是免疫原性低,能感染分裂相和非分裂相细胞,能自身携带片段整合入宿主细胞基因组,并在哺乳动物各类细胞稳定表达siRNA,长期抑制基因表达。技术实现要素:本发明提供一种靶向PLCε基因的RNA干扰重组病毒载体构建、筛选及其用途。本发明以RNA干扰技术为主,针对PLCε基因的不同靶序列,构建了四个能在293T细胞中表达siRNA的慢病毒真核表达载体pLv-PLCεshRNA,该载体是自身失活的慢病毒载体,其示意图谱见图1,能特异性的抑制PLCε基因表达。本发明的技术方案如下:一种靶向PLCε基因的siRNA重组慢病毒表达载体,是用于肿瘤PLCε基因相关性治疗性物质,慢病毒是由pLv-PLCεshRNA、pMD2.G和pSPAX2载体共转染293T细胞培养获得;其中,所述的pLv-PLCεshRNA重组载体是在plv-shRNA载体的多克隆位点中连接双链DNA片段构建而得,酶切位点是BamHI和EcoRI,所述的双链DNA片段为以下序列中的一种:(1)PLCε-homo-U1寡核苷酸序列1:正义链:5’GATCCCCACTTTGTTAGTCAGGAGATCTCGAGATCTCCTGACTAACAAAGTGGTTTTTG3’反义链:5’AATTCAAAAACCACTTTGTTAGTCAGGAGATCTCGAGATCTCCTGACTAACAAAGTGGG3’(2)PLCε-homo-U2寡核苷酸序列2:正义链:5’GATCCGCGGATTATTGGAACTCACTACTCGAGTAGTGAGTTCCAATAATCCGCTTTTTG3’反义链:5’AATTCAAAAAGCGGATTATTGGAACTCACTACTCGAGTAGTGAGTTCCAATAATCCGCG3’(3)PLCε-homo-U3寡核苷酸序列3:正义链:5’GATCCCCATCATTTATCATGGACATACTCGAGTATGTCCATGATAAATGATGGTTTTTG3’反义链:5’AATTCAAAAACCATCATTTATCATGGACATACTCGAGTATGTCCATGATAAATGATGGG3’(4)PLCε-homo-U4寡核苷酸序列4:正义链:5’GATCCGCTGCAATGTTTGAGGCAAATCTCGAGATTTGCCTCAAACATTGCAGCTTTTTG3’反义链:5’AATTCAAAAAGCTGCAATGTTTGAGGCAAATCTCGAGATTTGCCTCAAACATTGCAGCG3’根据本发明,所述的靶向PLCε基因的siRNA重组慢病毒表达载体的构建方法,包括步骤如下:a.根据PLCεmRNA序列,设计合成双链DNA片段,所述的双链DNA片段为所述的PLCε-homo-U1、PLCε-homo-U2、PLCε-homo-U3及PLCε-homo-U4核酸序列中的一种;b.然后将所述的双链DNA片段连接到plv-shRNA载体的多克隆位点中构建成pLv-PLCεshRNA重组载体;c.再将所述的pLv-PLCεshRNA重组载体与pMD2.G和pSPAX2载体共转染293T细胞培养,得靶向PLCε基因RNA干扰的重组慢病毒载体系统。为了对靶向PLCε基因RNA干扰的重组慢病毒载体对PLCεε基因的抑制效果进行鉴定,将pLv-PLCεεshRNA与包装质粒共转染293T细胞后进行荧光定量PCR检测目的基因表达水平,筛选出有效干扰质粒。附图说明图1为实施例中慢病毒表达载体plv-shRNA结果示意图。图2PLCε干扰RNA慢病毒载体瞬转293T细胞48h图片(荧光、可见光)。其中(a)为普通光镜(100×);(b)为荧光光镜(100×)。图3不同干扰序列下的PLCε表达水平。具体实施方式下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。主要材料:293T细胞购自中国科学院上海细胞生物学研究所;大肠杆菌感受态DH5α购自北京全式金生物技术有限公司;慢病毒载体plv-shRNA购自Clontech公司,含有人U6启动子,编码绿色荧光蛋白报告基因;各种限制性核酸内切酶、T4DNA连接酶及TaqDNA聚合酶均为美国NEB公司产品;SYBRGreenIqRT-PCRMixs购与上海生工生物工程技术服务公司;胎牛血清及DMEM培养液购自美国Gibico公司。实施例1靶向PLCε基因的慢病毒载体构建1.寡聚核酸的设计与合成设计靶向PLCε基因(NM_016341.3)的4个干扰序列(表1),并合成相应的单链DNA表1靶向PLCε基因RNA干扰序列序列名称序列信息起始位置U1CCACTTTGTTAGTCAGGAGAT965U2GCGGATTATTGGAACTCACTA2999U3CCATCATTTATCATGGACATA4343U4GCTGCAATGTTTGAGGCAAAT5479shRNA寡核苷酸的3’末端是TTTTT,作为RNA聚合酶Ⅲ型启动子U6的终止信号,其5’和3’末端分别是BamHI和EcoRI的限制性酶切位点,plv-shRNA模板中的loop结构选用了CTCGAG。各自单链DNA合成片段如下:(1)PLCε-homo-U1寡核苷酸序列1:正义链:5’GATCCCCACTTTGTTAGTCAGGAGATCTCGAGATCTCCTGACTAACAAAGTGGTTTTTG3’反义链:5’AATTCAAAAACCACTTTGTTAGTCAGGAGATCTCGAGATCTCCTGACTAACAAAGTGGG3’(2)PLCε-homo-U2寡核苷酸序列2:正义链:5’GATCCGCGGATTATTGGAACTCACTACTCGAGTAGTGAGTTCCAATAATCCGCTTTTTG3’反义链:5’AATTCAAAAAGCGGATTATTGGAACTCACTACTCGAGTAGTGAGTTCCAATAATCCGCG3’(3)PLCε-homo-U3寡核苷酸序列3:正义链:5’GATCCCCATCATTTATCATGGACATACTCGAGTATGTCCATGATAAATGATGGTTTTTG3’反义链:5’AATTCAAAAACCATCATTTATCATGGACATACTCGAGTATGTCCATGATAAATGATGGG3’(4)PLCε-homo-U4寡核苷酸序列4:正义链:5’GATCCGCTGCAATGTTTGAGGCAAATCTCGAGATTTGCCTCAAACATTGCAGCTTTTTG3’反义链:5’AATTCAAAAAGCTGCAATGTTTGAGGCAAATCTCGAGATTTGCCTCAAACATTGCAGCG3’2.寡聚核酸退火将合成的寡聚核酸分别用无酶水溶解,浓度为20μM。取相应正义链和反义链溶液,按照如下配比配制退火反应体系组分体积(μL)退火缓冲液5正义链(20μM)5反义链(20μM)5水至终体积50在PCR仪上按照如下程序进行退火处理:95℃5min;85℃5min;75℃5min;70℃5min;4℃保存。3.pl-shRNA载体线性化取2μgplv-shRNA载体,按照如下体系进行酶切处理:组分体积(μL)10×缓冲液5BamHI1EcoRI1水至终体积5037℃酶切1小时,1.5%琼脂糖电泳,使用DNA纯化试剂盒回收,核酸蛋白浓度测定仪测定回收质量。4.plv-PLCε-shRNA载体的构建利用T4DNA连接酶,按照如下体系进行载体连接反应:组分体积(μL)10×T4连接缓冲液2载体回收2shDNA2T4DNA连接酶1水至终体积2020℃连接过夜,转化质大抽杆菌感受态DH5α,挑选阳性克隆送去测序。本步骤得到的产物是plv-PLCε-shRNA。实施例2RNA干扰慢病毒质粒瞬转293T细胞将4种干扰慢病毒质粒,采用瞬时转染的方法转染293T细胞。293T细胞采用含10%FBS的高糖DMEM培养基,置于37℃,5%CO2培养箱内培养。转染前1天消化293T细胞,计数,将细胞接种至35mm培养皿内,每个培养皿接种6×105个细胞;次日用不含FBS的DMEM培养基分别配制钙转试剂和RNA干扰质粒,每84μl培养基内加入16μl钙转试剂,每100μl培养基内加入4μg质粒,分别混匀后将钙转溶液和质粒溶液轻轻混匀,室温静置10分钟,逐滴加入培养皿内,在37℃,5%CO2培养箱内培养24h,更换新鲜的含10%FBS的高糖DMEM培养基继续培养48h,收集细胞,提取细胞RNA。实施例31.总RNA的提取、cDNA的合成及RNA干扰效应的RT-PCR检测分别取1μgRNA反转录cDNA,采用荧光定量PCR法检测PLCε的表达水平,以beta-actin作为内参,荧光定量PCR所采用的引物序列见表2,采用2-△△CT相对定量方法计算不同RNA干扰序列样品的PLCε表达水平。筛选出表达水平最低的RNA干扰序列。检测结果(见图3)表明U4序列的PLCε基因表达水平最低,说明U4序列的RNA干扰效果最好。2.PLCε和beta-actin引物探针设计引物名称序列F-PLCεTCTTCGCACAATACCTACCTCACR-PLCεATCAATGGCTTCAACCACTTCCTF-actinbTATCGCCGCGCTCGTCGTCR-actinbTCTTGCTCTGGGCCTCGTCSEQUENCELISTING<110>同济大学苏州研究院<120>靶向PLCε基因RNA干扰重组慢病毒载体及其构建方法<130>2016<160>8<170>PatentInversion3.5<210>1<211>59<212>DNA<213>人工合成<400>1gatccccactttgttagtcaggagatctcgagatctcctgactaacaaagtggtttttg59<210>2<211>59<212>DNA<213>人工合成<400>2aattcaaaaaccactttgttagtcaggagatctcgagatctcctgactaacaaagtggg59<210>3<211>59<212>DNA<213>人工合成<400>3gatccgcggattattggaactcactactcgagtagtgagttccaataatccgctttttg59<210>4<211>59<212>DNA<213>人工合成<400>4aattcaaaaagcggattattggaactcactactcgagtagtgagttccaataatccgcg59<210>5<211>59<212>DNA<213>人工合成<400>5gatccccatcatttatcatggacatactcgagtatgtccatgataaatgatggtttttg59<210>6<211>59<212>DNA<213>人工合成<400>6aattcaaaaaccatcatttatcatggacatactcgagtatgtccatgataaatgatggg59<210>7<211>59<212>DNA<213>人工合成<400>7gatccgctgcaatgtttgaggcaaatctcgagatttgcctcaaacattgcagctttttg59<210>8<211>59<212>DNA<213>人工合成<400>8aattcaaaaagctgcaatgtttgaggcaaatctcgagatttgcctcaaacattgcagcg59当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1