一种筛选线粒体基因组编辑工具靶向目标序列的方法与流程
本发明属于基因组工程领域,具体地说,涉及一种筛选线粒体基因组编辑工具靶向目标序列的方法。
背景技术:
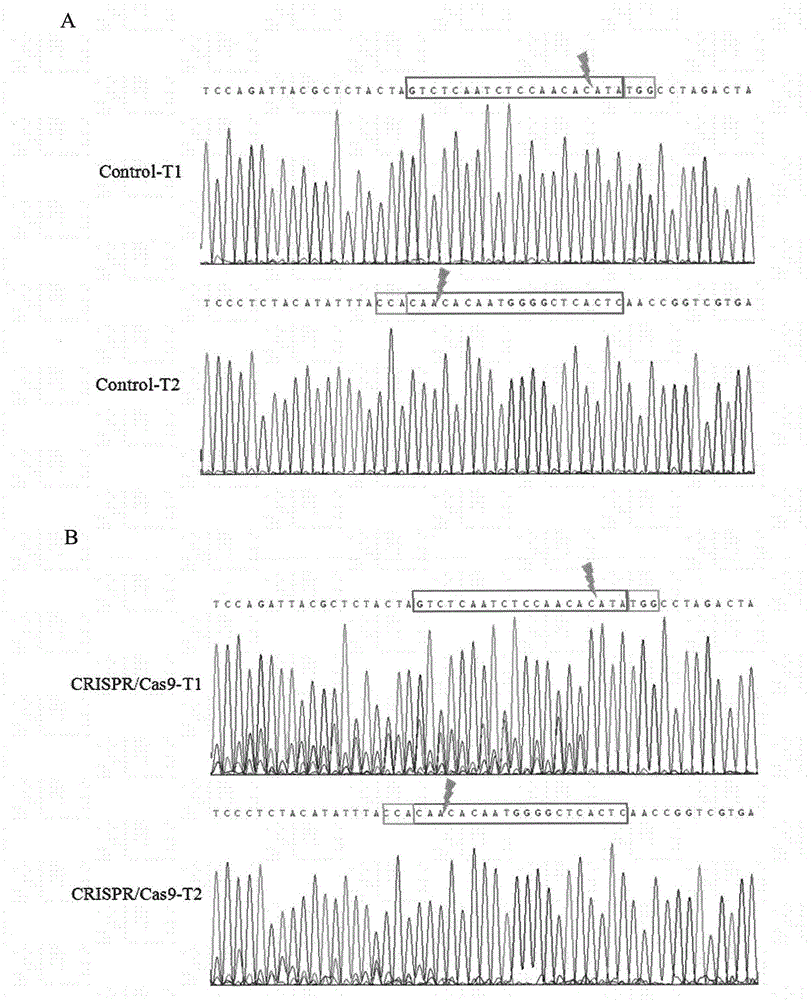
:随着测序技术的不断发展,我们获得越来越多的遗传信息。研究人员试图通过对特定靶向基因的破坏或敲除来研究它们的相关功能。与传统基因打靶技术相比,新型基因组编辑技术应用范围更广、作用效率更高、所需成本更低,可用于模式动物构建、基因功能研究、基因治疗等方面。目前,常用的基因组编辑技术主要包括以下三种:锌指蛋白(zincfingerproteins,ZFPs)技术、转录激活因子样效应因子(transcriptionactivator-likeeffectors,TALEs)技术、CRISPR(clusteredregularlyinterspacedshortpalindromicrepeats)技术。它们的大致原理都是通过造成靶向DNA双链断裂(double-strandbreaks,DSBs),引发细胞内固有的非同源末端连接(nonhomologousendjoining,NHEJ)或同源重组(homology-directedrepair,HDR)两种修复机制对DSBs处进行修复,引入插入或缺失突变。三种技术在具体操作方式上又有所不同:锌指核酸酶(zinc-fingernuclease,ZFN)与转录激活子样效应因子核酸酶(transcriptionactivator-likeeffectornuclease,TALEN)这两种人工核酸内切酶,均由DNA识别域与非特异性核酸内切酶FokI融合而成。其中,ZFN的锌指模块识别三联体碱基,在序列的识别上灵活性小,设计成本昂贵、技术被少数公司垄断,这些都限制了它的发展。TALEN在结构上较之ZFN更加优化。其DNA结合域中的各单元可与靶向目标序列一一对应,组装更为简单,特异性更高。2013年,一种源自细菌适应性免疫系统的CRISPR/Cas9的出现,为基因靶向修饰领域带来了巨大变革。它的主要原理:向导RNA(guideRNA,gRNA)与Cas9核酸酶形成的复合物,在gRNA指导下与靶向目标序列结合,由靶向目标序列下游的原型间隔序列相邻基序(protospaceradjacentmotifs,PAM)激活Cas9核酸酶,使其对PAM上游3nt处剪切造成DSBs。该系统较之前两种,应用范围更广、作用效率更高、使用成本更低,经过短短两年的发展,已被用于世界各地的实验室。线粒体是存在于大多数真核生物细胞中的细胞器,有“细胞中的能量工厂”之称。它主要通过氧化磷酸化以三羧酸循环腺苷(ATP)的形式为细胞活动提供能量。线粒体基因组,又称线粒体DNA(mitochondrialDNA,mtDNA)。长度一般为几万至数十万碱基对,人类mtDNA为16,569bps,共编码2个rRNA、13个多肽、22个tRNA。基因排列紧凑,除与mtDNA复制及转录有关的一小段区域外,无内含子序列。mtDNA表现为母系遗传,突变率高,是细胞核内DNA的10倍左右。且mtDNA突变是导致人类线粒体遗传病及衰老性疾病的重要原因之一。研究表明,它与衰老及神经退行性病变如阿尔茨海默症、帕金森氏病等老年化疾病有关,同时也与肿瘤的形成密切相关。截至目前,已鉴定出超过500多个致病性突变来自mtDNA,而针对mtDNA致病性突变的精确改造能力则相对缺乏。随着MichalMinczuk等将ZFN作用于mtDNA,SandraRBacman等首次将TALEN用于清除病人线粒体内的突变型mtDNA。这两种基于ZFPs技术、TALEs技术的线粒体基因组编辑工具都为更好的进行线粒体功能研究、线粒体遗传病的治疗等提供了可能。此外,本实验室也基于人工CRISPR/Cas9系统独特地构建了作用于mtDNA的mtCRISPR/Cas9系统,作为新型线粒体基因组编辑工具作用于mtDNA。任何一种基因组编辑技术,它的靶向剪切效率、切割特异性等问题不可回避。由于核基因组中的DNA损伤修复系统比较完整,当发生DSBs时,可通过NHEJ修复在靶向目标序列处引入插入或缺失突变。因此,靶向修饰核基因组的相关基因组编辑技术可通过错配酶法、靶向目标序列直接PCR后TA克隆测序或观察靶向目标序列测序峰图等方法鉴定。而相应的线粒体基因组编辑工具,由于线粒体缺乏完善的DNA损伤修复系统,修复能力较弱,无法通过上述方法检测。本发明,利用转基因技术将一段含有待选靶向目标序列的mtDNA序列片段或片段组合整合到宿主基因组中,获得转基因细胞株。通过相应基因组编辑技术靶向剪切活性的鉴定方法检测待选mtDNA靶向目标序列的剪切活性,从中筛选合适mtDNA靶向目标序列。建立了一种筛选线粒体基因组编辑工具靶向目标序列的方法。技术实现要素:本发明的目的在于,提供一种筛选线粒体基因组编辑工具靶向目标序列的方法。本发明基于转基因技术,构建了一个核基因组中含有线粒体DNA(mtDNA)序列的转基因细胞系。利用相应基因组编辑技术靶向剪切活性的鉴定方法检测待选mtDNA靶向目标序列的剪切活性,从中筛选合适mtDNA靶向目标序列。本发明提供了一种转基因细胞系的构建方法。所述转基因细胞系构建的主要流程:构建一个含有mtDNA序列的重组克隆质粒,将该质粒转染于作用细胞后(或线性化后转染),经适宜浓度的药物筛选(或其它方法),使这段mtDNA序列随机整合到宿主基因组中,挑选单拷贝或低拷贝的单克隆转基因细胞株用于检测待选mtDNA靶向目标序列的剪切活性,从中筛选合适mtDNA靶向目标序列。所述转基因细胞系的构建,可将一个或多个待选mtDNA靶向目标序列、潜在脱靶序列、定点整合所需同源臂序列或其它序列片段(或片段组合)连入克隆载体质粒中,并将这段序列整合到宿主基因组中,构建转基因细胞系。所述克隆载体质粒,可以为哺乳动物细胞表达载体pEGFP-N1,也可以为其它真核表达载体。所述重组克隆质粒的构建,可通过一步或多步常规PCR扩增得到一段含有一个或多个待选mtDNA靶向目标序列和其它序列片段(或片段组合)的串联序列,将其连入克隆载体质粒中,若该载体有荧光标签,可同时与其中的荧光标签(EGFP、RFP等)编码序列融合,经测序验证,得正确的重组克隆质粒。优选地,该克隆载体质粒可为哺乳动物细胞表达载体pEGFP-N1,购自Clontech公司,含neo抗性基因和EGFP标签,可在多克隆位点(MCS)处插入外源基因并于EGFP融合,由CMV启动子启动,由于含有荧光标签可通过荧光显微镜观察绿色荧光情况,检测融合蛋白的表达情况。所述以pEGFP-N1克隆载体质粒为例,将一段含有两个mtDNA靶向目标序列T1、T2的序列连入其中的重组克隆质粒构建流程如图1A所示:1、以HEK293基因组DNA为模板,经CL-F1/CL-R1、CL-F2/CL-R2、CL-F3/CL-R3三对引物依次扩增得一段同时含有T1、T2靶向目标序列的mtDNA序列;2、将上述序列由NheI/AgeI两位点,经双酶切连入,经测序得正确质粒;3、以上述步骤所得正确质粒为模板,经CL-F4/CL-R4引物对扩增去除EGFP编码序列前的起始密码子ATG,由AgeI/NotI经双酶切连入,通过测序得到含mtDNA靶向目标序列融合基因的正确重组克隆质粒pEGFP-mtDNA。所述重组克隆质粒pEGFP-mtDNA中外源基因表达模块如图1B所示,核苷酸序列如SEQIDNO.1所示。其中,CMV启动子的核苷酸序列如SEQIDNO.1中第1-589位核苷酸所示;NheI限制性酶切位点的核苷酸序列如SEQIDNO.1中第591-596位核苷酸所示;KozaK序列的核苷酸序列如SEQIDNO.1中第597-602位核苷酸所示;起始密码子的核苷酸序列如SEQIDNO.1中第603-605位核苷酸所示;T1靶向目标序列的核苷酸序列如SEQIDNO.1中第636-655位核苷酸所示;T2靶向目标序列的核苷酸序列如SEQIDNO.1中第752-771位核苷酸所示;AgeI限制性酶切位点的核苷酸序列如SEQIDNO.1中第773-778位核苷酸所示;编码EGFP的核苷酸序列如SEQIDNO.1中第780-1493位核苷酸所示;终止密码子的核苷酸序列如SEQIDNO.1中第1494-1496位核苷酸所示;NotI限制性酶切位点的核苷酸序列如SEQIDNO.1中第1498-1505位核苷酸所示。所述鉴定用单拷贝或低拷贝转基因细胞株的挑选,可首先通过常规PCR、蛋白质印迹法(WesternBlot)等方法初筛阳性单克隆细胞株,再经实时荧光定量PCR法、SouthernBlot法或其它方法检测外源基因拷贝数。优选地,阳性单克隆细胞株的初筛可通过常规PCR法完成。优选地,外源基因拷贝数的检测可通过实时荧光定量PCR法完成。所述常规PCR法初筛阳性单克隆细胞株,以所述构建的重组克隆质粒pEGFP-mtDNA为例,主要流程:1、将pEGFP-mtDNA质粒转染于培养在60mm细胞培养皿中的作用细胞中;2、24小时后,将细胞传代至10cm细胞培养皿中同时开始药筛;3、每隔3-4天更换一次含有相同浓度抗生素的新鲜培养基,至细胞不再大批量死亡,且形成清晰单克隆细胞团后,挑取单克隆于24孔板中;4、收取含绿色荧光的细胞,同时提取基因组,以此为模板分别用P1(P1-F/P1-R)、P2(P2-F/P2-R)、P3(P3-F/P3-R)三对引物进行常规PCR初筛,均可扩增出目的条带的细胞为初筛所得阳性单克隆细胞株。所述P1、P2、P3引物对,根据pEGFP-mtDNA序列设计,扩增范围涵盖启动子区、编码区、终止区。其中,P1-F位于CMV启动子区、P1-R位于EGFP编码序列上、P2-F位于CMV启动子区、P2-R位于EGFP编码序列上(含终止密码子)、P3-R位于PolyA的编码序列上,基于pEGFP-N1构建的重组克隆质粒普遍适用;P3-F位于外源基因序列上(含起始密码子ATG),可根据实际情况自行设计。所述实时荧光定量PCR法检测外源基因拷贝数,以所述构建的重组克隆质粒pEGFP-mtDNA为例,主要流程:1、以ΔCt(ΔCt=Ct特异-Ct内参)和标准样品拷贝数的对数值(以2为底)作图得绝对定量标准曲线;2、提取初筛阳性单克隆细胞株基因组,利用实时荧光定量PCR检测其对应的ΔCt值;3、将各细胞株的ΔCt值带入标准曲线对应的计算公式中,得拷贝数。所述绝对定量标准曲线建立流程:1、将重组克隆质粒与作用细胞的基因组DNA混合,分别设置含有1、2、4、8、16个外源基因拷贝数的标准品对照(若重组克隆质粒abp,作用细胞基因组DNA用量bng、大小cbp,则bng基因组DNA中含有一个外源基因拷贝数的质量为a×b×0.5/cng,该法为通用方法);2、以上述标准品为样品,利用实时荧光定量PCR,经RT-F/RT-R、Actin-F/Actin-R(内参)两引物对扩增,得ΔCt;3、由ΔCt值和对应标准样品拷贝数的对数值(以2为底)作图得标准曲线。所述外源基因拷贝数的检测流程:1、提取初筛所得细胞株的基因组DNA,以此为模板,经RT-F/RT-R、Actin-F/Actin-R(内参)两引物对扩增,得ΔCt;2、将各个细胞株对应的ΔCt值带入标准曲线所得公式中,计算相应拷贝数。所述实时荧光定量PCR法起始模板量小、灵敏性高、简单高效,足以满足本发明的筛选需要。两引物对RT-F/RT-R(127bp)、Actin-F/Actin-R(130bp),扩增大小相近、Tm值基本相等、扩增效率基本相同。本发明还提供了一种检测mtDNA靶向目标序列剪切活性的方法。所述mtDNA靶向目标序列剪切活性检测的主要流程:将含有相应mtDNA靶向目标序列的基因组编辑技术作用质粒转染到所构建的单拷贝或低拷贝转基因细胞株中,经合适作用时间,利用相应基因组编辑技术靶向剪切活性的鉴定方法进行检测。所述基因组编辑技术靶向剪切活性的鉴定方法主要有:1、对靶向目标序列进行PCR扩增,扩增产物经变性、退火后形成错配,通过错配酶剪切不完全匹配的DNA序列,验证靶向目标序列剪切活性;2、对靶向目标序列进行PCR扩增后,通过TA克隆测序与理论序列比对验证靶向目标序列剪切活性;3、对靶向目标序列进行PCR扩增后,扩增产物直接测序,通过观察靶位点处的套峰情况验证靶向目标序列剪切活性,等多种方式进行检测。优选地,基因组编辑技术靶向剪切活性的鉴定方法可通过观察PCR产物的测序峰图检测。所述观察测序峰图检测靶向目标序列剪切活性的原理:如果靶向目标序列在核基因组中发生DSBs,则会引发细胞中的NHEJ修复机制,得到插入或缺失突变。而对样品全基因组DNA进行PCR扩增所得的扩增产物为混合产物,其中既含有野生型原始靶向目标序列,又含有引入插入或缺失的突变型靶向目标序列。因此,查看靶向剪切处是否有套峰出现,如有套峰,则证明靶向目标序列有剪切活性,反之则无。所述通过观察测序峰图检测mtDNA靶向目标序列剪切活性,以pEGFP-mtDNA所构建的单拷贝或低拷贝转基因细胞系为例,主要流程:1、将含有相应mtDNA靶向目标序列的基因组编辑技术作用质粒转染到所构建的单拷贝或低拷贝转基因细胞株中;2、48-72小时后,收取细胞提基因组DNA,以此为模板经P1引物对扩增,扩增产物送样测序;3、观察测序峰图,若靶向剪切位置处有套峰出现,则证明靶向目标序列有剪切活性,反之则无。本发明的一种筛选线粒体基因组编辑工具靶向目标序列的方法,具有如下优点:所述重组克隆质粒在构建时,研究者可以将一个或多个待选mtDNA靶向目标序列、潜在脱靶序列、定点整合所需同源臂序列或其它序列片段(或片段组合)连入克隆载体质粒中,并将这段序列整合到宿主基因组中,构建单拷贝或低拷贝的转基因细胞系。该细胞系不仅可以检测多个待选靶向目标序列的剪切活性,还可以比较不同靶向目标序列的剪切效率、分析单碱基或多碱基不匹配下的潜在脱靶效应等问题,为更好的筛选靶向目标序列、设计线粒体基因组编辑工具提供重要的参考依据和实验基础,一举多得。所述挑选的单拷贝或低拷贝单克隆转基因细胞株,在转入定量作用质粒后,可以更加清晰的检测待选靶向目标序列剪切活性,减少背景干扰。所述检测方法适用范围广,基于ZFN、TALEN、CRISPR/Cas9等基因组编辑技术改造而成的线粒体基因组编辑工具,或其它线粒体基因组编辑工具均可使用此方法检测靶向目标序列的剪切活性,比较不同靶向目标序列间的剪切效率,筛选合适靶向目标序列,简单高效。附图说明图1为本发明中pEGFP-mtDNA重组克隆质粒构建示意图。其中,图1A为pEGFP-mtDNA重组克隆质粒的构建流程图,红色方块表示mtDNA靶向目标序列T1、T2,蓝色方块表示flag标签;图1B为pEGFP-mtDNA重组克隆质粒中外源基因表达模块示意图,红色部分表示mtDNA靶向目标序列T1、T2,蓝色部分表示flag标签,黄色部分表示T1、T2分别对应的PAM,绿色部分表示EGFP。图2为本发明中的单克隆转基因细胞株初筛结果。其中,图2A为pEGFP-mtDNA质粒转染效率图;图2B为单克隆细胞株常规PCR初筛结果图。图3为本发明利用绝对定量PCR法检测外源基因拷贝数。其中,图3A为特异性引物对RT-F/RT-R的扩增曲线与熔解曲线;图3B为内参引物对Actin-F/Actin-R的扩增曲线与熔解曲线;图3C为ΔCt与拷贝数的对数值(log2N)作图得标准曲线,Log2N=-1.1772ΔCt+2.7793(R2=0.9971)。图4为本发明中T1、T2两个靶向目标序列剪切活性的检测。其中,图4A为不含mtDNA靶向目标序列T1、T2的CRISPR/Cas9空骨架质粒,即Control-T1、Control-T2,转染于转基因细胞株10#,48小时后的测序峰图;图4B为将含mtDNA靶向目标序列T1、T2的CRISPR/Cas9质粒,即CRISPR/Cas9-T1、CRISPR/Cas9-T2,转染于转基因细胞株10#,48小时后的测序峰图;红色方框表示靶向目标序列,黄色方框表示PAM,灰色闪电状图标指示Cas9核酸酶剪切位置(PAM上游3nt处)。具体实施方式以下通过具体实施例对本发明进一步说明,但并不限定本发明。在本发明基础上,所做的同类型改进均属于本发明要求保护的范围。实施例1单克隆转基因细胞株的获得本发明提供了一种转基因细胞株,其构建流程:将含有mtDNA靶向目标序列的重组克隆质粒瞬时转染于细胞中,经药物筛选,挑单克隆于24孔板。提取单克隆细胞株的基因组DNA,通过常规PCR初筛得阳性单克隆细胞株,再利用实时荧光定量PCR挑选其中单拷贝或低拷贝的单克隆转基因细胞株。1.1构建重组克隆质粒:以pEGFP-N1为载体,将一段同时含有两个靶向目标序列(T1、T2)的mtDNA序列与EGFP的N端融合,除去EGFP前的起始密码子ATG,最终得到重组克隆质粒:pEGFP-mtDNA(如图1所示)。具体步骤如下:1)以HEK293基因组为模板,用CL-F1/CL-R1引物对进行PCR扩增(335bp),得产物J;2)对J进行胶回收,并以此为模板用CL-F2/CL-R2(CL-F2带有flag标签,如表1横线部分所示)引物对进行PCR扩增(167bp),得产物K;3)对K进行胶回收,并以此为模板用CL-F3/CL-R3(CL-F3带有NheI酶切位点和flag标签、CL-R3带有AgeI酶切位点,如表1相应横线、横线斜体部分所示)引物对进行PCR扩增(194bp),得产物L;4)对纯化后的产物L及pEGFP-N1质粒做NheI/AgeI双酶切,分别以此为插入片段及载体进行连接、转化、挑单克隆鉴定、鉴定正确样品测序;5)以上一步骤中的正确质粒为模板,用CL-F4/CL-R4(CL-F4带有AgeI酶切位点,如表1横线斜体部分所示;删去EGFP起始密码子ATG,添加一个碱基C调整编码框,该碱基的选择应避免引入终止密码子)引物对进行PCR扩增(902bp),得产物M;6)对纯化后的产物M及上一步骤所用质粒做AgeI/NotI双酶切,分别以此为插入片段及载体进行连接、转化、挑单克隆鉴定、鉴定正确样品测序,即得最终质粒:pEGFP-mtDNA。重组克隆质粒的构建流程如图1A所示,其中的外源基因表达模块如图1B所示。上述具体步骤并不限定本发明,在构建重组克隆质粒时,可根据需要将一个或多个待选靶向目标序列、潜在脱靶序列等同时串联入质粒中,便于同时检测其剪切活性,筛选合适靶向目标序列。在外源基因与EGFP的N端融合时要注意是否需要调整编码框,是否引入了起始密码子或终止密码子等问题。相关引物序列具见表1:1.2初筛阳性单克隆细胞株:具体步骤如下:1)转染前一天,将HEK293细胞接种于60mm细胞培养皿中;2)第二天,将1.1中所述鉴定正确的重组克隆质粒pEGFP-mtDNA瞬时转染于HEK293细胞中,24小时后观察EGFP的表达情况(转染效率达95%以上,如图2A所示),并将细胞传代至10cm细胞培养皿中;3)加800ug/ml的G418筛选阳性细胞,每隔3-4天更换一次含有相同浓度抗生素的新鲜培养基;4)培养至细胞不再大批量死亡,且形成清晰单克隆细胞团后,挑取单克隆于24孔板中继续培养;5)待24孔板长满后,将其中11个含有绿色荧光的单克隆细胞传代至6孔板中扩大培养并收取部分细胞提基因组(基因组提取试剂盒购自天根生化科技(北京)有限公司,货号DP304-03);6)以步骤5)中所述11个基因组样品为模板,分别用P1(P1-F/P1-R)、P2(P2-F/P2-R)、P3(P3-F/P3-R)三对引物(相关序列如表2所示)进行常规PCR初筛,三对引物均可扩增出目的条带的细胞,则为初筛所得阳性单克隆细胞株,即编号为2#、3#、5#、8#、10#的5株细胞(如图2B所示)。所述步骤6)中,三对引物P1、P2、P3均根据pEGFP-mtDNA序列设计,扩增范围涵盖启动子区、编码区、终止区,保证筛选结果准确性的同时又确保了外源基因能够完整表达。具体扩增范围如下:1)P1:CMV部分+mtDNA靶向目标序列+EGFP部分(967bp);2)P2:CMV小部分+mtDNA靶向目标序列+EGFP(1074bp);3)P3:mtDNA靶向目标序列+EGFP+polyA部分(1069bp)。相关引物序列具见表2:引物名称引物序列(5’-3’)对应IDP1-FATGTTCCCATAGTAACGCCAASEQIDNO.10P1-RGGGTCTTGTAGTTGCCGTCGSEQIDNO.11P2-FGATTTCCAAGTCTCCACCCCSEQIDNO.12P2-RTCACTTGTACAGCTCGTCCATGSEQIDNO.13P3-FATGGACTACAAGGACGACGATGSEQIDNO.14P3-RTTTGTGATGCTATTGCTTTATTTGSEQIDNO.151.3挑选单拷贝或低拷贝的单克隆转基因细胞株:经过药物筛选得到的阳性单克隆细胞株,其外源基因往往随机整合在染色体上。因此,在经1.2所述方法初筛阳性单克隆细胞株后,检测其外源基因拷贝数,挑选单拷贝或低拷贝的单克隆转基因细胞株是有效鉴定靶向目标序列剪切活性的关键步骤。所述检测外源基因拷贝数的流程:1)以ΔCt(ΔCt=Ct特异-Ct内参)和标准样品拷贝数的对数值(以2为底)作图得绝对定量标准曲线;2)提取初筛阳性单克隆细胞株基因组并以此为模板,通过实时荧光定量PCR得各细胞株的ΔCt值;3)将各细胞株的ΔCt值带入标准曲线对应的计算公式中,得拷贝数(N);1.3.1建立绝对定量标准曲线:具体步骤如下:1)将pEGFP-mtDNA质粒与HEK293基因组DNA混合,分别设置含有1、2、4、8、16个外源基因拷贝数的标准品对照(假设质粒大小为abp,HEK293基因组DNA用量为bng,HEK293基因组DNA大小为3×109bp,则bng基因组DNA中含有一个外源基因拷贝数的质量为a×b×0.5/3×109ng);2)设计特异性扩增外源基因序列(RT-F/RT-R)的引物对和扩增内参β-actin(Actin-F/Actin-R)的引物对(引物序列具见表3);3)利用实时荧光定量PCR,以步骤1)中5个不同拷贝数的标准品为样品,经RT-F/RT-R、Actin-F/Actin-R(内参)两引物对扩增,每个反应重复3次,取平均Ct值。所述实时荧光定量PCR的反应总体系20ul(上/下游引物各0.5ul、ddH2O9ul、2×SYBRpremixE×Taq10ul),反应程序:95℃3min;95℃15S、60℃30S(40个循环),熔解曲线60℃-95℃,每个样品做3次重复。结果如图3所示,特异性引物对RT-F/RT-R的扩增曲线、熔解曲线如图3A所示,内参引物对Actin-F/Actin-R的扩增曲线、熔解曲线如图3B所示,两熔解曲线均在86℃附近有且只有一个单峰,证明两引物对均为特异性扩增。将RT-F/RT-R扩增的CtpEGFP-mtDNA值减去Actin-F/Actin-R扩增的CtActin值,得ΔCt,ΔCt与标准品拷贝数的对数值(log2N)作图得到标准曲线(如图3C所示),Log2N=-1.1772ΔCt+2.7793,决定系数R2=0.9971。相关引物序列具见表3:引物名称引物序列(5’-3’)对应IDRT-FCACAGCCCTATACTCCCTCTACATSEQIDNO.16RT-RTTACGTCGCCGTCCAGCSEQIDNO.17Actin-FAACGGTGAAGGTGACAGCAGTSEQIDNO.18Actin-RCTTCCTGTAACAACGCATCTCATASEQIDNO.191.3.2检测单克隆转基因细胞株的拷贝数:具体步骤如下:1)提取1.2初筛所得2#、3#、5#、8#、10#5株细胞的基因组;2)利用实时荧光定量PCR,以上述所提基因组为模板,经RT-F/RT-R、Actin-F/Actin-R(内参)两引物对扩增,得ΔCt;3)将5个细胞株对应的ΔCt值带入Log2N=-1.1772ΔCt+2.7793公式中,计算相应拷贝数(N)。结果如表4所示,标准差在0.9351和0.1387之间,结果可信。挑选单拷贝的细胞株10#用于检测线粒体基因组编辑工具靶向剪切活性。外源基因拷贝数检测结果具见表4:克隆编号拷贝数(N±S)2#2.08±0.93513#1.39±0.66805#1.36±0.13878#5.70±0.514910#1.03±0.1948实施例2待选mtDNA靶向目标序列的筛选本发明还提供了一种筛选线粒体基因组编辑工具靶向目标序列的方法,主要流程:将含有相应待选mtDNA靶向目标序列的基因组编辑技术作用质粒转染到所选转基因细胞株中。48-72小时后,收取细胞提基因组,并以此为模板,用P1-F/P1-R引物对PCR扩增。扩增产物送样测序,测序引物为P1-R。观察测序峰图,若靶向剪切位置处有套峰出现,则证明靶向目标序列有剪切活性,反之则无。根据待选mtDNA靶向目标序列的测序峰图套峰强弱,比较其剪切活性,从中筛选合适靶向目标序列。具体步骤如下:1)构建分别含有mtDNA靶向目标序列T1、T2的两个CRISPR/Cas9质粒,命名为CRISPR/Cas9-T1、CRISPR/Cas9-T2;2)将CRISPR/Cas9-T1、CRISPR/Cas9-T2和相应不含T1、T2靶向目标序列的CRISPR/Cas9空骨架质粒(Control-T1、Control-T2)分别瞬时转染到1.3.2所述的转基因细胞株10#中;3)48小时后,收取细胞提基因组,以此为模板用P1-F/P1-R引物对进行PCR扩增,扩增产物送样测序。检测结果如图4所示,阴性对照Control-T1、Control-T2在靶向目标序列处均无套峰出现(图4A),而作用质粒CRISPR/Cas9-T1、CRISPR/Cas9-T2在靶向目标序列处均有套峰出现,且CRISPR/Cas9-T1处的套峰强于CRISPR/Cas9-T2处(图4B)。证明两个靶向目标序列T1、T2均有剪切活性,且T1的剪切效率高于T2。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1