一种整合缺陷型慢病毒载体、其制备方法及应用与流程
本发明涉及医学生物技术领域,尤其涉及一种整合缺陷型慢病毒载体、其制备方法及应用。
背景技术:
基因治疗技术在当代越来越具有主要地位。但是基因治疗的技术手段有很大的局限。而慢病毒载体的出现有望突破常规载体的瓶颈,慢病毒载体是目前基因治疗与基因递送中应用最广泛有效的工具。
以1型人免疫缺陷病毒(HIV-1)为基础构建的慢病毒载体具有可感染非分裂细胞、免疫反应小、携带的基因片段容量大和可整合进宿主基因组而长期表达等优点,慢病毒载体能够容纳多达8kb的外源基因,在体内体外均能高效转导包括DC细胞在内广泛的细胞类型,能够整合外源基因到宿主基因组中引起持续的基因表达,且没有预存免疫,载体骨架的免疫原性弱,因而成为最理想的基因转移载体之一。
慢病毒载体在体内应用,其主要挑战是转基因整合入宿主基因组所引起的安全问题。传统的慢病毒三质粒(第1个质粒为含有HIV病毒gag基因和pol基因的包装质粒;第2个为转基因质粒,含有HIV包装信号ψ,反转录及整合所需要的长末端重复序列(LTR)和报告基因;第3个质粒表达包膜蛋白)共转染许可细胞(293FT),许可细胞提供了病毒增殖装配的基本环境条件,颗粒的形成主要靠三质粒系统完成。
然而,慢病毒的随机性整合到靶细胞染色体中也会促使细胞基因组的一些功能基因被沉默或者引起原癌基因插入激活致细胞癌变等严重负面影响。作为体内应用,其主要挑战是转基因整合入宿主基因组所引起的安全问题。
整合型慢病毒载体的应用不但避免了随机整合导致插入诱变致癌的危险,而且保持了慢病毒载体的其它优点。此外,在整合慢病毒载体的基础上通过转座子引导的定点整合机制可以进一步改造成定点整合慢病毒载体。
技术实现要素:
针对现有技术的不足及实际的需求,本发明提供一种整合慢病毒载体,,证实其整合缺陷特性,大大提高慢病毒载体应用的安全性,为临床应用奠定基础。
为达此目的,本发明采用以下技术方案:
一方面,本发明提供一种整合缺陷型慢病毒载体,所述慢病毒载体在pCMV-dR8.91慢病毒质粒的4846-4848位碱基编码的苯丙氨酸突变为丙氨酸和/或在pCMV-dR8.91慢病毒质粒的4328-4830位碱基编码的组氨酸突变为丙氨酸。
具体地,所述慢病毒载体只在pCMV-dR8.91慢病毒质粒的4846-4848位碱基编码的苯丙氨酸突变为丙氨酸,或
所述慢病毒载体只在pCMV-dR8.91慢病毒质粒的4328-4830位碱基编码的组氨酸突变为丙氨酸,或
所述慢病毒载体在pCMV-dR8.91慢病毒质粒的4846-4848位碱基编码的苯丙氨酸和4328-4830位碱基编码的组氨酸同时突变为丙氨酸。
本发明中,慢病毒载体在pCMV-dR8.91慢病毒质粒的4846-4848位碱基编码的苯丙氨酸突变为丙氨酸或pCMV-dR8.91慢病毒质粒的4328-4830位碱基编码的组氨酸突变为丙氨酸将其中一位氨基酸进行突变,酶活会下降,而将这两个位置的氨基酸同时进行突变,酶活大大降低。
优选地,所述苯丙氨酸突变为丙氨酸是通过pCMV-dR8.91慢病毒质粒的4846-4848位的碱基由CA突变为GC。
优选地,所述组氨酸突变为丙氨酸是通过pCMV-dR8.91慢病毒质粒的4828-4830位的碱基由TT突变为GC。
本发明中,pCMV-dR8.91慢病毒质粒的4846-4848位的碱基和4828-4830位的碱基突变后的序列如SEQ ID NO.7所示。
第二方面,本发明提供一种重组慢病毒,如第一方面所述的慢病毒载体与pFUGW表达载体及包膜载体pVSV-G载体进行共转染哺乳细胞得到的重组慢病毒。
第三方面,本发明提供一种宿主细胞,包括如第一方面所述的慢病毒载体或如第二方面的重组慢病毒;
优选地,所述宿主细胞为293T细胞。
第四方面,本发明提供一种如第一方面所述的慢病毒载体的制备方法,包括如下步骤:
(1)根据序列设计引物,以pCMV-dR8.91慢病毒质粒为模板进行扩增,得到一个突变的基因片段;
任选地,(2)将步骤(1)扩增得到的基因片段作为模板进行扩增,得到具有两个突变的基因片段;
(3)将步骤(1)或步骤(2)扩增得到的基因片段连接到pCMV-dR8.91慢病毒质粒中,构建重组质粒;
(4)将重组质粒转化大肠杆菌DH5α,筛选阳性克隆;
其中,所述步骤(2)可选择性的进行,即只突变一个位点时直接将步骤(1)突变的基因片段连接到pCMV-dR8.91慢病毒质粒中,若同时突变两个位点时,再进行步骤(2),将步骤(2)突变的基因片段连接到pCMV-dR8.91慢病毒质粒中。
优选地,步骤(1)所述引物为SEQ ID NO.1-6所示的序列。
所述SEQ ID NO.1所示的核苷酸序列的上游引物如下:cagaacgtctcagtaccagttagagaaagaacccata;
所述SEQ ID NO.2所示的核苷酸序列的下游引物如下:tactgtgatatttctcagcttcttcttgggcct;
所述SEQ ID NO.3所示的核苷酸序列的上游引物如下:aggcccaagaagaagctgagaaatatcacagta;
所述SEQ ID NO.4所示的核苷酸序列的下游引物如下:ccttttcttttaagcttgtggatgaatactg;
所述SEQ ID NO.5所示的核苷酸序列的上游引物如下:cagtattcatccacaagcttaaaagaaaagg;
所述SEQ ID NO.6所示的核苷酸序列的下游引物如下:ttgcagaattccatgtgttaatcctcatc.
优选地,步骤(2)扩增的引物为SEQ ID NO.1和SEQ ID NO.6所示的序列。
所述SEQ ID NO.1所示的核苷酸序列的上游引物如下:cagaacgtctcagtaccagttagagaaagaacccata;
所述SEQ ID NO.6所示的核苷酸序列的上游引物如下:ttgcagaattccatgtgttaatcctcatc.
优选地,步骤(3)所述将扩增得到的基因片段连接到pCMV-dR8.91慢病毒质粒的Acc65I和EcoRI克隆位点。
第五方面,本发明提供一种如第一方面所述的慢病毒载体或如第二方面所述的重组慢病毒在制备基因治疗药物中的应用。
与现有技术相比,本发明具有如下有益效果:
(1)本发明制备的突变后的pCMV-dR8.91慢病毒载体酶活性大大丧失,但不影响转导效率,更安全更高效;
(2)本发明制备的突变后的pCMV-dR8.91慢病毒载体具有整合性,同整合型慢病毒对比,整合型慢病毒颗粒在第6天还有85%的活性,而本发明的整合缺陷型慢病毒颗粒只有30%;到第21天时,整合型慢病毒颗粒有80%的活性,而本发明的整合缺陷型慢病毒颗粒已经没有活性了。
附图说明
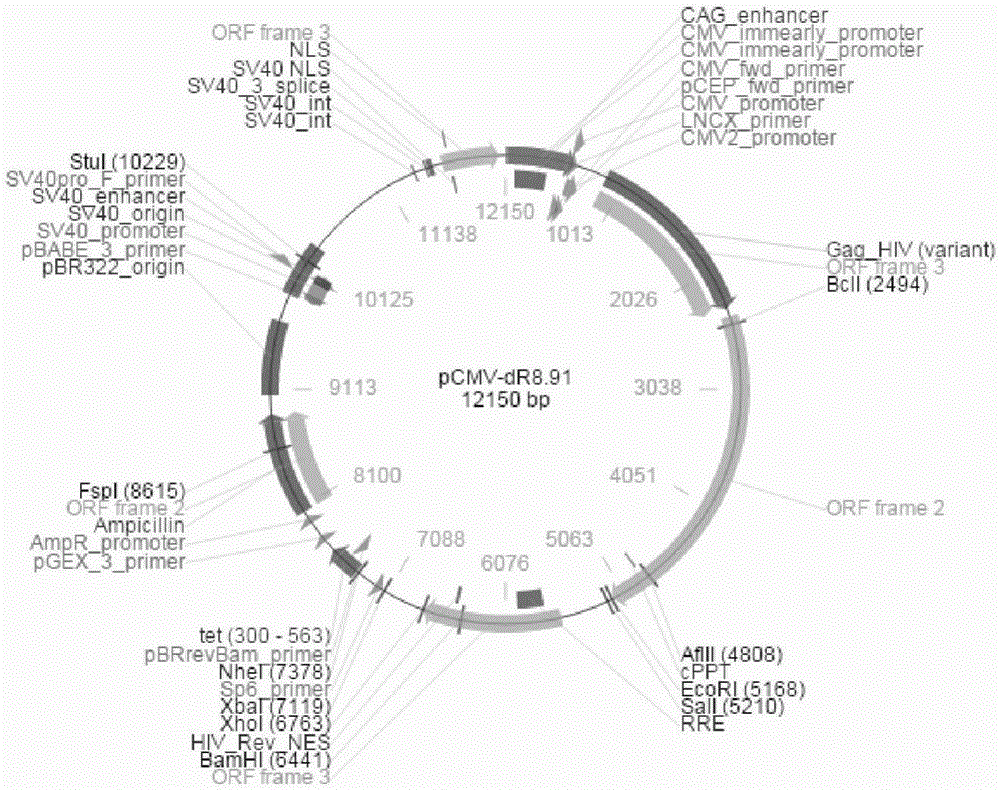
图1是本发明pCMV-dR8.91慢病毒载体图谱;
图2是本发明pCMV-dR8.91慢病毒载体4328-4329位的碱基突变的序列图;
图3是本发明pCMV-dR8.91慢病毒载体4846-4848位的碱基突变的序列图;
图4是本发明pCMV-dR8.91慢病毒载体的4328-4329位的碱基突变阳性克隆测序比对结果;
图5是本发明pCMV-dR8.91慢病毒载体的4846-4848位的碱基突变阳性克隆测序比对结果;
图6是本发明pCMV-dR8.91慢病毒载体PCR二次点突变后的扩增条带;
图7是本发明pCMV-dR8.91慢病毒载体在Acc65I/EcoRI限制性内切酶的作用下切成的328+950+10.8K 3个片段;
图8是本发明慢病毒感染细胞的GFP表达百分比和平均荧光强度,其中,为野生型,为突变型。
具体实施方式
为更进一步阐述本发明所采取的技术手段及其效果,以下结合附图并通过具体实施方式来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
实施例1:构建突变后的pCMV-dR8.91慢病毒载体
1)pCMV-dR8.91慢病毒载体及要突变的位点,如图1和图2所示:
(1)根据已知pCMV-dR8.91慢病毒载体序列设计引物如下:
上游引物:cagaacggtctcagtaccagttagagaaagaacccata(SEQ ID NO.1);
下游引物:tactgtgatatttctcagcttcttcttgggcct(SEQ ID NO.2);
上游引物:aggcccaagaagaagctgagaaatatcacagta(SEQ ID NO.3);
下游引物:ccttttcttttaagcttgtggatgaatactg(SEQ ID NO.4);
上游引物:cagtattcatccacaagcttaaaagaaaagg(SEQ ID NO.5);
下游引物:ttgcagaattccatgtgttaatcctcatc(SEQ ID NO.6)
以pCMV-dR8.91慢病毒载体为模板,直接用于第一个突变位点的PCR扩增。
PCR扩增反应体系如下:
其中,引物的浓度为1OD的引物溶于400μL ddH2O,引物为SEQ ID NO.1-2、SEQ ID NO.3-4和SEQ ID NO.5-6同时进行PCR,总共设置三组反应组,一组以水为样本的阴性对照。
反应条件如下:
所获得PCR产物进行1%的琼脂糖凝胶电泳鉴定并用胶回收试剂盒回收纯化。将得到的PCR产物再进行第二轮扩增。以第一轮PCR产物为模板,直接用于第二个突变位点的PCR扩增。
PCR扩增反应体系如下:
其中,引物的浓度为1OD的引物溶于400μL ddH2O,设置一组以水为样本的阴性对照。
反应条件如下:
获得PCR产物进行1%的琼脂糖凝胶电泳鉴定并用胶回收试剂盒回收纯化,结果如图6所示,扩增条带的大小为1278kb。
2)酶切连接pCMV-dR8.91慢病毒质粒中,构建重组质粒,反应体系如下:
37℃恒温水浴锅中反应3h,酶切产物经胶纯化回收,pCMV-dR8.91慢病毒载体在Acc65I/EcoRI限制性内切酶的作用下切成的328+950+10.8K 3个片段如图7所示。
将酶切后的载体与基因进行连接,反应体系如下:
轻轻混匀组分,置于16℃水浴锅反应2h,得到重组载体。
(2)将重组载体转化DH5α感受态细胞,挑选阳性克隆进行测序验证,结果如图4-5所示,克隆序列与原始序列完全一致。
实施例2:慢病毒的包装与滴定
(1)将测序获得的目的突变载体即为pCMV-dR8.91m,然后将pCMV-dR8.91,pCMV-dR8.91m分别与pFUGW表达载体及包膜载体pVSV-G载体进行共转染293T细胞,转染后48-72h收上清,上清中分别含有整合型慢病毒颗粒FGR和整合缺陷型慢病毒颗粒FGRm;
(2)将上清3000rpn,离心10min,取上清,用0.45μm的滤器进行过滤;
(3)取100uL检测感染滴度,取200uL进行拷贝数检测,取16mL进行无菌检测,取1mL进行内毒检测,将剩余的液体,进行100000G超速离心2h,沉淀用相应的培养基重悬。
得到的重悬液可直接进行后续实验或-70℃保存,用BIOMERIEUX的Vironostika HIV-1抗原微量ELISA检测试剂盒进行测定,取慢病毒收获上清,按产品说明书进行p24抗原定量,同时进行病毒滴度的测定,经过调整为1×109TU/mL。
实施例3:整合缺陷型慢病毒颗粒的整合特性分析
按1×107接种293T细胞于25cm2小方瓶中,次日将同等p24含量的FGR(整合型慢病毒颗粒)和FGRm(整合缺陷型慢病毒颗粒)分别感染293T细胞,感染后会出现GFP的表达,同时设正常细胞对照,感染后的第3、6、9、12和15天等用流式细胞术比较整合和整合慢病毒颗粒感染细胞的GFP表达百分比和平均荧光强度,如图8所示,可以看出,整合型慢病毒颗粒在第6天还有85%的活性,而本发明的整合缺陷型慢病毒颗粒只有30%;到第21天时,整合型慢病毒颗粒有80%的活性,而本发明的整合缺陷型慢病毒颗粒已经没有活性。
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!