一种即配即用型纳米靶向药物载体的制备方法与流程
本发明属于纳米材料技术领域,具体涉及一种在纳米粒子表面通过即时组装修饰功能分子的方法,以满足即时装配即时使用(即配即用)型靶向药物载体的需求,尤其是满足个性化精准治疗的需求。
背景技术:
纳米材料的使用在我们的生活中已经非常普遍,目前各种类型的纳米粒子均可方便制备,如有机聚合物纳米粒子,无机纳米粒子,有机/无机杂化纳米粒子等。通常情况下,在纳米粒子表面接上一些修饰分子会使之带有一些新的特定功能,比如药物纳米载体粒子表面接上靶向分子就会使其具有靶向相应肿瘤细胞的能力,接上光声分子就会使其具有光声成像的能力等等。这些修饰进一步拓展了纳米粒子在生物医学等方面的应用潜力。
目前在纳米粒子上修饰功能性分子的方法,通常是在纳米粒子表面先修饰官能团,再用可与之进行化学反应的功能性分子通过化学键接的方式修饰在纳米粒子表面,若要得到纯净的修饰功能分子的纳米粒子,通常还需要进行一系列的分离纯化操作以及进行冷冻干燥操作等。这种方法能按照要求在纳米粒子上修饰所需的功能分子,然而通常耗时较长,步骤繁琐,所以很难满足在现实工作中根据不同的要求即刻得到所需纳米粒子并立即投入使用的需求。随着技术水平的快速发展和人们对于个性化精准治疗的日益增长需求,这种方法已经不能满足纳米粒子个性化装配以及即时使用的要求。
技术实现要素:
本发明针对技术背景中所提及的问题,提供一种普适的、在纳米粒子表面修饰功能分子的方法,使得修饰的步骤更加简便,以满足即时装配即时使用(即配即用)型靶向药物载体的需求,尤其是满足个性化精准治疗的需求。
本发明提供的即配即用型纳米靶向药物载体的制备方法,利用链霉亲和素(SA)对生物素(BT)有非常高的亲和力,且反应呈高度专一性的特点,将已经制备好的BT修饰的功能分子固定在修饰了SA的纳米粒子表面上。所使用的条件温和,修饰过程方便简洁,并且能够达到即时装配即时使用的特点。具体步骤如下:
第一步,在纳米粒子的表面接枝上链霉亲和素(SA),制备得到表面有SA的纳米粒子;
第二步,制备功能分子修饰的生物素(BT);
第三步,将SA表面修饰的纳米粒子和一种或多种功能分子修饰的BT,通过亲和素-生物素之间高强度的亲和作用,得到表面功能分子修饰的纳米粒子,无需任何后处理即可将其立即用于后续的应用试验中。
其中,第一步的具体操作步骤为:
将0.1~2 份表面带有羧基/羟基/氨基的纳米粒子(这里的“/”为“或”的意思)、0.05~1 份1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.05~1份N-羟基琥珀酰亚胺(NHS)分散在10-200 份缓冲溶液中,活化0.5~12 小时后再加入0.1~2 份链霉亲和素;搅拌反应12~24小时;用离心的方式将没有反应的SA和小分子反应物去除,并用PBS反复洗多次,得到SA接枝的纳米粒子;
第二步的具体操作步骤为:
将0.05~1份功能分子放入缓冲溶液中,加入0.1~2 份生物素,并用0.05~1 份1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)和0.05-1 份1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)活化,搅拌反应12~24小时;用分子量为1000的透析袋透析2~3天;得到带有不同功能分子的生物素衍生物(BT);
第三步的具体操作步骤为:
将0.05~0.5 份第一步得到的表面有链霉亲和素的纳米粒子分散在缓冲溶液当中,添加一定质量比的一种或者多种第二步得到的功能分子修饰的生物素(BT),在10~25℃下混合,即获得一种或多种表面修饰功能分子的纳米粒子,并将其立即用于后续应用试验中。
在此基础上,还可以将得到的表面修饰功能分子的纳米粒子再次分散在缓冲溶液当中,添加入一定质量比的另一种或者多种功能分子修饰的生物素(BT),在10~25℃即时混合,即可获得另一种或多种表面修饰功能分子的纳米粒子立时投入后续应用试验中。
本发明的目标是得到即时装配即时使用(即配即用)型靶向药物载体,以满足个性化精准治疗的需求。
本发明中,混合功能分子和纳米粒子一般只需1~5分钟。
本发明中,第一步中所使用的链霉亲和素来自阿维丁链霉菌或是来自鸡蛋白的亲和素。
本发明中,第二步中所使用的生物素可以是D-生物素,生物素酰肼等,其功能分子的反应官能团基团可以为氨基、羧基或羟基。生物素和功能分子的选择遵循以下原则:若生物素的反应官能团为羧基时,则功能分子反应基团必须为羟基或者氨基;若生物素的反应官能团为羟基或者氨基时,则功能分子反应基团必须为羧基。
本发明中,第一步中所使用的纳米粒子可以是表面带有上述所要求基团的磁性纳米晶簇、磁流体、二氧化硅纳米粒子、二氧化钛纳米粒子、聚合物纳米粒子或重金属纳米粒子等。此处只是列举部分纳米粒子,还包括未列出的其他适合本发明的纳米粒子。
本发明中,第二步中,所使用的功能分子可以是异硫氰酸酯荧光素(FITC)、叶酸、罗丹明、RGD。此处只是例举一些常见的功能性分子,还包括未列出的其他适合本发明的所有带可反应官能团的功能分子。
本发明中,第三步中,功能分子修饰的BT和SA的纳米粒子的质量比可以为1:1000~1:1。优选1:600~1:50。
本发明中,第三步中,混合后的纳米粒子不经过任何后处理即刻应用到后续的应用中,后续的实验可以是细胞实验,也可以是其他检测或者标记实验。
本发明中,所使用的缓冲溶液可以是磷酸氢二钠/磷酸二氢钠缓冲体系、硼酸/硼酸钠缓冲体系、柠檬酸/柠檬酸钠缓冲体系、醋酸/醋酸钠缓冲体系(这里的“/”为“和”的意思),且pH值可以调节在6~8。
本发明中,所有的原料易得,合成步骤简单,条件温和,在纳米粒子的修饰结束后无需进行任何后处理可直接用于下一步的试验,很好的满足了现代科技对于即时组装效率的诉求,提供了极大的便利性,为拓宽纳米粒子的使用范围打下的坚实的基础。
附图说明
图1、实施例1的磁性纳米晶簇上修饰叶酸的荧光光谱图。
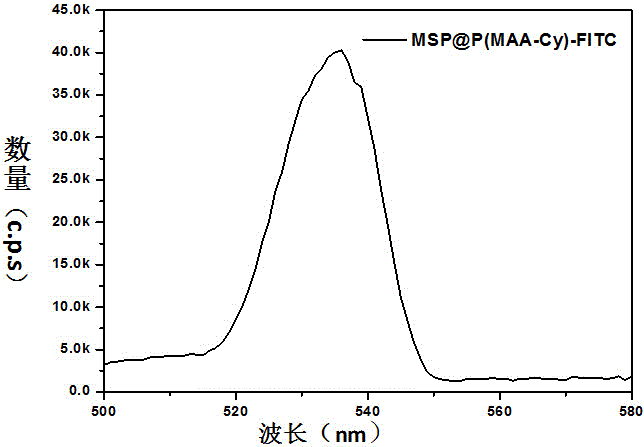
图2、实施例2的磁性纳米晶簇上修饰FITC的荧光光谱图。
图3、实施例3的磁性纳米晶簇上修饰罗丹明的荧光光谱图。
图4、实施例4的磁性纳米晶簇上同时修饰罗丹明、叶酸和FITC的荧光光谱图。
图5、实施例6的载药磁性纳米晶簇上修饰靶向分子在肿瘤SK-OV-3细胞中的流式图。其中,a为12小时后,叶酸、RGD修饰的载药磁性纳米晶簇和叶酸,RGD同时修饰的载药磁性纳米晶簇被SK-OV-3细胞摄取的情况并与后处理过的载药纳米粒子最对比,b为24小时后的。
图6、实施例7的载药磁性纳米晶簇上修饰靶向分子对于肿瘤SK-OV-3细胞的毒性实验图并与后处理过的载药纳米粒子作对比。其中,(a)DOX浓度为1μm/ml以及48小时后对于细胞的毒性试验图,(b)DOX浓度为5 μm/ml以及48小时后对于细胞的毒性试验图,(c)DOX浓度为10 μm/ml以及48小时后对于细胞的毒性试验图,(d)DOX浓度为1μm/ml以及72小时后对于细胞的毒性试验图,(e)DOX浓度为5 μm/ml以及72小时后对于细胞的毒性试验图,(f)DOX浓度为10 μm/ml以及72小时后对于细胞的毒性试验图。
图7、本发明方法流程图示。
具体实施方式
下面通过具体实施例进一步描述本发明。
实施例1:通过SA和BT的强相互作用,在磁性纳米晶簇表面修饰叶酸分子的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后用真空干燥的方法在40℃下干燥2天,即得到SA表面修饰的磁性纳米晶簇。
将0.1g叶酸放入水中,加入0.05 g的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.05g N-羟基琥珀酰亚胺(NHS),在25oC下活化2h;然后加入0.2g生物素,在25℃搅拌反应12h。最后得到叶酸修饰的生物素。
最后将0.1 g第一步得到的SA接枝的磁性纳米晶簇分散在缓冲溶液中,添加10 mg的叶酸修饰的BT,在25℃下充分混合,无需任何处理,即得到表面修饰叶酸的磁性纳米晶簇。
实施例2:通过SA和BT的强相互作用,在磁性纳米晶簇表面修饰FITC分子的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后用真空干燥的方法在60 ℃下干燥1天,即得到SA表面修饰的磁性纳米晶簇。
将0.1gFITC放入水中,然后加入0.2g生物素酰肼,在25℃搅拌反应24 h,最后得到FITC修饰的生物素。
最后将0.1 g第一步得到的SA接枝的磁性纳米晶簇分散在缓冲溶液当中,添加10 mg的FITC修饰的BT,在10℃下充分混合,无需任何处理,即得到表面修饰FITC的磁性纳米晶簇。
实施例3:通过SA和BT的强相互作用,在磁性纳米晶簇表面修饰罗丹明分子的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后用真空干燥的方法在50℃下干燥1.5天,即得到SA表面修饰的磁性纳米晶簇。
将0.1g罗丹明放入水中,加入0.05 g的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.05g N-羟基琥珀酰亚胺(NHS),在35oC下活化10h,然后加入0.2g生物素,在35℃搅拌反应18 h,最后得到罗丹明修饰的生物素。
最后将0.1 g第一步得到的SA接枝的磁性纳米晶簇分散在缓冲溶液当中,添加10 mg的罗丹明修饰的BT,在15℃下充分混合,无需任何处理,即得到表面修饰叶酸的磁性纳米晶簇。
实施例4:通过SA和BT的强相互作用,在磁性纳米晶簇表面同时修饰罗丹明、FITC和叶酸分子的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后用真空干燥的方法在45℃下干燥2天,即得到SA表面修饰的磁性纳米晶簇。
将0.1g罗丹明放入水中,加入0.05 g的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.05g N-羟基琥珀酰亚胺(NHS),在50℃下活化20小时,然后加入0.2g生物素,在30℃搅拌反应20 h,最后得到罗丹明修饰的生物素。叶酸修饰的BT的制备方法参见实施例1;FITC修饰的BT的制备方法参见实施例2。
最后将0.1g第一步得到的PEG接枝的磁性纳米晶簇分散在缓冲溶液当中,添加10 mg的罗丹明修饰的BT、10 mg的FITC修饰的BT和10 mg的叶酸修饰的BT,在30℃下充分混合,无需任何处理,即得到表面修饰罗丹明、FITC和叶酸的磁性纳米晶簇。
实施例5:通过SA和BT的强相互作用,在磁性纳米晶簇表面修饰靶向分子RGD的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后用真空干燥的方法在55℃下干燥1天,即得到SA表面修饰的磁性纳米晶簇。
将0.1gRGD放入水中,加入0.05 g的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.05g N-羟基琥珀酰亚胺(NHS),在40℃下活化10h,然后加入0.2g生物素,在40℃搅拌反应10 h,最后得到靶向分子RGD修饰的生物素。
最后将0.1 g第一步得到的SA接枝的磁性纳米晶簇分散在缓冲溶液当中,添加10 mg的RGD修饰的BT,在20℃下充分混合,无需任何处理,即得到表面修饰RGD的磁性纳米晶簇。
实施例6:通过SA和BT的强相互作用,在负载DOX药物的磁性纳米晶簇表面修饰靶向分子并即时使用在流式细胞实验的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后在磁性纳米晶簇上负载18%的DOX,用真空干燥的方法在45℃下干燥1.5天,即得到SA表面修饰的负载DOX药物的磁性纳米晶簇。
叶酸修饰的BT的制备方法参见实施例1,RGD修饰的BT的制备方法参见实施例5。将0.1 g第一步得到的SA接枝的磁性纳米晶簇分散在缓冲溶液当中,添加10 mg的RGD修饰的BT,在10℃下充分混合,无需任何处理,即得到表面修饰RGD的载药磁性纳米晶簇。添加10 mg的叶酸修饰的BT,在10℃下充分混合,无需任何处理,即得到表面修饰叶酸的载药磁性纳米晶簇。同时添加10 mg的叶酸修饰的BT和10 mg的叶酸修饰的BT,在10℃下充分混合,无需任何处理,即得到表面同时修饰叶酸和RGD的载药磁性纳米晶簇。
修饰后,立即将DOX 浓度为2μg/ml并修饰了靶向分子的载药磁性纳米晶簇混入培养基中与SK-OV-3肿瘤细胞培养,12小时和24小时后测试其进入细胞的数量。数据表明即时使用样品和经过纯化等后处理的样品进入细胞的效果基本一致。
实施例7:通过SA和BT的强相互作用,在负载DOX药物的磁性纳米晶簇表面修饰靶向分子并即时使用于靶向杀伤癌细胞的方法
将0.1g表面带有羧基的磁性纳米晶簇(MSP),0.1g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和0.1gN-羟基琥珀酰亚胺(NHS)分散在100 ml磷酸缓冲溶液中,活化两个小时后再加入0.2 g链霉亲和素。搅拌反应24小时后,用磁分离的方式将没有反应的SA和小分子反应物去除,并反复磁分离5次,之后在磁性纳米晶簇上负载18%的DOX,用真空干燥的方法在60℃下干燥1天,即得到SA表面修饰的负载DOX药物的磁性纳米晶簇。
叶酸修饰的BT的制备方法参见实施例1,RGD修饰的BT的制备方法参见实施例5。将0.1 g第一步得到的SA接枝的磁性纳米晶簇分散在缓冲溶液当中,添加10 mg的RGD修饰的BT,在25℃下充分混合,无需任何处理,即得到表面修饰RGD的载药磁性纳米晶簇。添加10 mg的叶酸修饰的BT,在25℃下充分混合,无需任何处理,即得到表面修饰叶酸的载药磁性纳米晶簇。同时添加10 mg的叶酸修饰的BT和10 mg的叶酸修饰的BT,在25℃下充分混合,无需任何处理,即得到表面同时修饰叶酸和RGD的载药磁性纳米晶簇。
修饰后,立即将DOX 浓度为1,5,10μg/ml修饰靶向分子的载药磁性纳米晶簇混入培养基中与SK-OV-3肿瘤细胞培养,48小时和72小时后测试其杀伤癌细胞的效果。数据表明即时使用样品和经过纯化等后处理的样品杀伤癌细胞的效果基本一致。
- 还没有人留言评论。精彩留言会获得点赞!
- 靶向PD-1的多肽及其应用的制造方法与工艺
- 靶向同位素生产系统的制造方法与工艺
- 在制备68GA‑螯合物官能化的靶向剂中用作金属抑制剂的单糖、二糖或多糖的制造方法与工艺
- 用于引导和靶向按需物质递送的纳米结构的载体的制造方法与工艺
- 通过PROSTRATIN靶向K‑RAS介导的信号转导通路和恶性肿瘤的制造方法与工艺
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺