水溶性酸响应淋巴靶向缓释载体纳米炭、制备方法及应用与流程
本发明属于肿瘤靶向示踪及治疗领域,具体涉及一种水溶性酸响应淋巴靶向药物缓释载体纳米炭及其制备方法,还涉及由该纳米炭和抗肿瘤药物制备得到的药剂及其制备方法,以及涉及该纳米炭制备淋巴示踪和肿瘤治疗制剂方面的用途和所得药剂在制备淋巴示踪和肿瘤治疗药物方面的用途。
背景技术:
癌症是危害人体健康和威胁人类生命的元凶之一,肿瘤的难治在于它的多发、复发和转移。在肿瘤的整个发展历程中,肿瘤的转移和传播占据了关键的地位,也是癌症会成为人类最难攻克的疾病的主要原因之一。肿瘤的转移与传播是一个复杂的过程,对于不同的肿瘤来说,科学家至今未能对于它们的共性进行确切地阐述。有的肿瘤单单只依靠血液转移途径,有的只依靠淋巴转移途径,而有的在它们发展转移的不同阶段同时依赖着这两种途径。然而对于很多最常见和最致命的实体肿瘤的传播来说,淋巴转移往往发挥着最主要的作用,特别是胃癌和大部分的皮源性肿瘤,如乳腺癌、肺癌、结肠癌和前列腺癌等。
最近有研究发现在实体瘤中或实体瘤周存在有淋巴管,细胞因子和生长因子刺激着肿瘤淋巴并且肿瘤转移程度与肿瘤内新生淋巴管的程度有直接的关系,肿瘤细胞通过新生毛细淋巴管进入淋巴结,淋巴转移在癌症分期与预后中扮演了重要的角色。
因此临床上迫切需要一种能在术中清楚地显示肿瘤周边淋巴结的指示剂,定向地指示出已存在癌细胞转移的淋巴结,从而有助于彻底切除转移的淋巴结,防止癌症的转移和肿瘤的复发。
目前,较为常用的指示剂为活性炭混悬液。临床通常使用的纳米炭混悬注射液(商品名:卡纳琳,CNS)是通过特殊加工,将普通活性炭球磨制成粒径为21nm左右的光滑活性炭纳米颗粒(activated carbon nanoparticles,ACNPs),再辅以助悬剂聚乙烯毗咯烷酮(PVP),用生理盐水制成黑色混悬剂,形成平均粒径约150nm的纳米炭团。卡纳琳注射于肿瘤周围组织间隙后,纳米炭粒会迅速地以渗透和扩散的方式通过局部缺如或者不完整的毛细淋巴管的基底膜、内皮细胞间隙,以及巨噬细胞的吞噬和胞饮作用,主动运输或被动运载到局部淋巴管和淋巴结内,并部分滞留于引流淋巴结(黑染)。其余部分继续移行至下一站引流淋巴结,使区域内各站引流淋巴结形成黑染。
申请号为02113731.5的中国专利详细的公布了纳米炭混悬注射液的制备方法。然而,该专利所得的注射液需要大量的助悬辅料,其中作为辅料的聚乙烯吡咯烷酮,据美国FDA的资料显示,其可能影响凝血功能。同时,用于给药化疗时,该注射剂在使用前的配制和储藏受到许多限制性条件的约束,不利于临床使用。另外,该注射剂由于需要考虑到整个注射液体系的分散性,人们难以根据实际需求,向其中加入对于治疗而言重要甚至是必要的药物,极大的限制了其实际应用。
申请号为200410092028.9的中国专利改善了上述注射液的水溶性问题,其通过利用浓硝酸对纳米炭进行氧化,制备得到了无需分散助悬辅料的接枝纳米炭混悬液。
然而,人们发现仅对纳米炭进行氧化处理,静置时间久后,容易沉淀产生结块,重分散性不好,需进行超声处理才能很好的将纳米炭重新分散。这就意味着,在实际的医用中,必须(至少是很有必要)进行现配现用,否则一旦出现团聚甚至是结块则不能产生相应的淋巴示踪效果,给临床带来了极大不便。
另外,在临床中,虽然转移的肿瘤可被所用纳米炭染色,便于医生发现并将其切除或做其它相应处理,然而,当肿瘤过小以至于难以被医生识别,或者当转移的肿瘤处于难以进行相应手术的位置时,纳米炭则丧失了相应的临床意义,极大的影响了治疗效果。
对于难以利用手术或者常规剂型的药物进行治疗的肿瘤,通常的做法是将药物装载在具有肿瘤靶向性的药物载体上。该方法的一般要求是所用载体对药物的载药量高、药物的释放行为好以及具有一定的环境响应性。虽然目前人们已经获得了不少符合上述要求的肿瘤靶向药物载体,但是在临床实际中,为了完全清除转移的肿瘤,使用纳米炭的同时还使用其他靶向治疗药物无疑成本巨大。更重要的是,医疗人员完全可以仅仅选用治疗效果好的靶向治疗药物,这使得纳米炭的临床应用的必要性大为降低。
综上所述,为了提高纳米炭的临床使用价值,必须至少解决现有技术的如下缺点:(1)纳米炭容易结块且不易重分散;(2)现有纳米炭未能实现肿瘤治疗意义上的载药。
为了克服上述载药问题,中国专利200510021728.3通过将抗癌前药与纳米炭偶联,实现了将药物靶向释放至肿瘤区域的效果。然而,该专利没有解决纳米炭的重分散问题;同时适用的药物较少,需为具有活性较好的氨基、亚氨基或羟基的抗癌药物;另外其制备步骤和耗时较长且反应过程中容易造成抗癌药物的浪费。
因此,本领域亟需能同时解决上述现有技术缺点且易于应用的技术。
技术实现要素:
针对现有技术的缺点,本发明的目的之一在于提供一种水溶性酸响应淋巴靶向药物缓释载体纳米炭,该纳米炭由表面羧基含量为0.10-1.00mmol/g的活性炭与氨基聚乙二醇单甲醚接枝而得,接枝量为1.00-10.00%(W%),所述纳米炭的粒径为50-500nm。
如本发明的实施例所示,本发明纳米炭相对于仅仅经过氧化处理的纳米炭而言,具有更好的水分散性,这可能是由于PEG溶于水中,会在颗粒表面溶解伸展形成一定的空间体积排斥,从而增加了活性炭纳米颗粒在水溶液中的稳定性,并且减少了其发生聚沉的可能性。更重要的是,本发明的纳米炭静置之后不会形成结块,静置后的沉淀仅需稍加震荡便可重新分散。相比于商用产品卡纳琳而言,本发明纳米炭无需助分散剂,避免了PVP对人体凝血功能的潜在威胁。相比于中国专利200410092028.9所得产品而言,本发明纳米炭避免了静置后结块的产生并具有十分良好的重分散性,十分利于实际临床应用。
值得指出的是,本发明在解决现有技术存在的重分散性差的缺点之外,还解决了载药的问题。
中国专利200510021728.3提供的技术方案虽然实现了载药功能,但其成本高昂且受限于特定的抗癌药物,更重要是的其载药率受限于药物与纳米炭的接枝偶联程度,无法获得较好的载药率。另外,在携载如5-氟尿嘧啶这样常用的抗肿瘤药时,本发明纳米炭的药效可几乎达到纯药的药效。
如本发明的实施例所示,本发明可以获得很好的载药率的同时还能实现优秀的缓释效果,更重要的是,本发明在释药行为上还具有酸度响应性,当处于微酸环境(肿瘤区域)时,药物释放速度会相应加快。
通过上述说明,本领域人员不难理解,本发明实质上解决了限制纳米炭淋巴示踪剂临床应用的技术问题。具体而言,利用本发明纳米炭可以避免PVP对人体凝血功能造成影响的风险,也无需担忧由于静置时间久后产生结块导致纳米炭失去淋巴靶向功能。本发明除了保留了纳米炭的淋巴示踪功能,同时,对于无法示踪、不易于手术切除处理(或其它处理方式)的淋巴管和淋巴结,本发明可通过携载的药物进行治疗,无需过于担忧癌细胞转移和肿瘤复发。
值得指出的是,本发明无需进行一些认为是必要的环境响应性载体的有意设计,便获得了较好的酸度响应性。另外,本发明的释药行为是十分良好的缓释。本领域人员应当很容易理解,本发明上述特点可以使得本发明纳米炭作为优秀的肿瘤淋巴靶向药物载体。然而相比于一般的淋巴示踪制剂而言,本发明纳米炭可以对容易进行手术切除的淋巴进行示踪,也可以对不易观察到的或手术切除的淋巴管进行药物治疗,使得肿瘤的临床治疗更为高效。
本发明所述表面羧基含量为0.10-1.00mmol/g的活性炭是由活性炭经氧化剂氧化而得。
本发明所述氧化剂包括硝酸、双氧水、硫酸、臭氧中的至少一种,优选为双氧水。
双氧水作为氧化剂较为温和,且后处理较为方便,更宜于本发明的实施。
优选的,所述活性炭的表面羧基含量为0.60-1.00mmol/g。
所述氨基聚乙二醇单甲醚的分子量为500-5000Da,优选为2000Da。
优选的,所述接枝度为6.00-10.00%(W%)。
优选的,所述纳米炭的粒径为250-350nm。
本发明的另一个目的在于提供利用上述本发明纳米炭为药物载体的用于淋巴示踪和肿瘤治疗的药剂,该药剂包括所述纳米炭和抗肿瘤药物。
所述抗肿瘤药物包括5-氟尿嘧啶(5-fluorouracil,5FU)、奥沙利铂(Oxaliplatin,Oxi)、丝裂霉素C、卡波霉素、阿克拉霉素、氨甲喋呤、阿霉素、阿柔比星、紫杉醇、卡铂中的至少一种。
所述抗肿瘤药物为5-氟尿嘧啶和/或奥沙利铂。
优选的,所述抗肿瘤药物与所述纳米炭的重量比为5-20:100。
所述药剂为药学意义上可接受的任一剂型。
优选的,所述药剂的剂型为粉剂。
优选的,所述药剂还包括药学意义上可用的佐剂。
本发明的另一个目的在于提供制备上述纳米炭的方法,该方法包括如下步骤:
(1)将活性炭颗粒研磨至粒径为50-500nm;
(2)利用氧化剂对步骤(1)所得物进行氧化处理,所述氧化剂为硝酸、双氧水、硫酸、臭氧中的至少一种;
(3)将氨基聚乙二醇单甲醚通过活化脂缩合反应接枝到步骤(2)所得物上,控制接枝量为6.00-10.00%,即得。
优选的,所述氧化剂为双氧水,氧化处理方法为:称取活性炭纳米颗粒,先按1mL/g·活性炭纳米颗粒的比例加入超纯水,再在慢搅拌下,滴加1mL/g·活性炭纳米颗粒的双氧水,反应0.5h;然后升高温度至70℃,逐滴加入4mL/g·活性炭纳米颗粒的双氧水,控制3h滴完,滴加完后再充分反应2h;最后进行抽滤,用超纯水洗涤多次至pH呈中性,普通烘箱中先于60℃烘3h,再于120℃烘干至恒重。
优选的,步骤(3)中的接枝反应为:称取1g步骤(2)所得活性炭纳米颗粒于,加入100mL超纯水,在磁力搅拌下分散1h,再分别加入1.00mmol的1-乙基-3-(3-二甲氨丙基)碳二亚胺和1.00mmol的N-羟基丁二酰亚胺,室温反应1h,最后滴加入氨基聚乙二醇单甲醚水溶液,继续常温反应8h。
本发明的另一个目的在于提供利用上述本发明纳米炭制备用于淋巴示踪和肿瘤治疗的药剂的方法,该方法包括如下步骤:
(1)制备抗肿瘤药物水溶液,所述抗肿瘤药物包括5-氟尿嘧啶、奥沙利铂、丝裂霉素C、卡波霉素、阿克拉霉素、氨甲喋呤、阿霉素、阿柔比星、紫杉醇、卡铂中的至少一种;
(2)向步骤(1)所得物加入所述纳米炭,进行充分的超声震荡处理;
(3)将步骤(2)所得物制备成药学意义上可接受的剂型。
优选的,超声震荡处理时,温度为室温,处理时间为180min。
优选的,所述剂型为粉剂。
本发明的另一个目的在于提供本发明所述纳米炭在制备淋巴示踪和肿瘤治疗制剂方面的用途。
本发明的另一个目的在于提供本发明所述药剂在制备淋巴示踪和肿瘤治疗方面药物的用途,其特征在,所述肿瘤包括胃癌、结直肠癌、乳腺癌、大肠癌或头颈部癌。
本发明的主要有益效果:
1、本发明所得纳米炭具有十分优秀的水分散性和重分散性,克服了现有技术重分散性差的缺点;
2、本发明所得纳米炭可作为优秀的抗癌药物载体,具有载药量高、缓释效果好和酸度相应性的优点;
3、本发明易于实施,成本低廉;
4、本发明克服了现有技术中纳米炭在临床应用的缺点,具有十分良好的应用前景。
附图说明
图1为未经处理(左)和经过氧化处理(右)的TEM结果图;
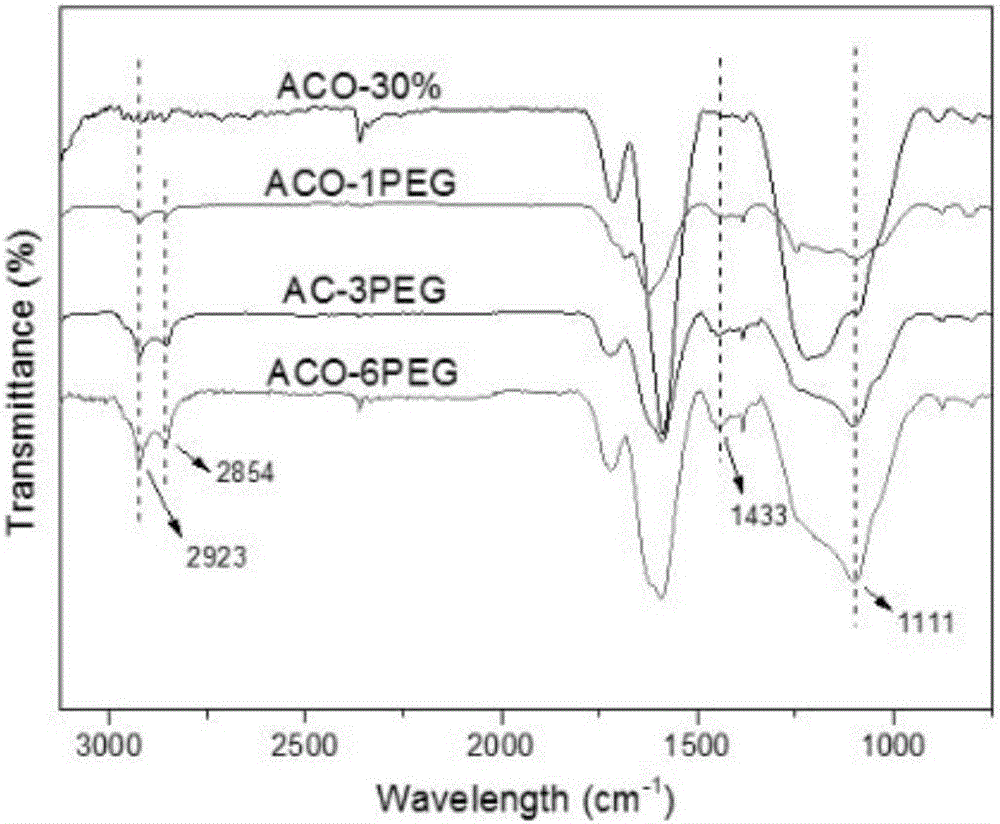
图2为不同纳米炭的FTIR结果图,其中,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-1PEG、ACO-2PEG和ACO-6PEG代表不同接枝度的本发明的纳米炭;
图3为不同纳米炭的TGA结果图,其中,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-1PEG、ACO-2PEG和ACO-6PEG代表不同接枝度的本发明的纳米炭;
图4中左图为不同纳米炭的粒径分布结果图,其中,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-1PEG、ACO-2PEG和ACO-6PEG代表不同接枝度的本发明的纳米炭;右图为本发明纳米炭的TEM结果图;
图5为不同纳米炭静置4天后的稳定性结果图,其中,AC-5为未经处理的纳米炭,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-6PEG代表本发明的纳米炭;
图6为不同纳米炭对5FU的载药量结果图;其中,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-1PEG和ACO-6PEG代表不同接枝度的本发明的纳米炭;
图7为不同纳米炭对5FU的释放结果图,其中,ACO-30%-5FU代表仅仅经过氧化处理所得的纳米炭对5FU的释放结果,ACO-6PEG-5FU代表本发明纳米炭对5FU的释放结果;
图8为不同纳米炭的CLSM结果图,其中,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-6PEG代表本发明的纳米炭;
图9为不同纳米炭包载FITC后的细胞内吞实验结果图,其中,ACO-30%代表仅仅经过氧化处理所得的纳米炭,ACO-6PEG代表本发明的纳米炭;
图10为不同纳米炭的MTT实验结果图,AC-5-5FU为未经处理的携载5FU的纳米炭,ACO-30%-5FU代表仅仅经过氧化处理所得的携载5FU的纳米炭,ACO-6PEG-5FU代表本发明的携载5FU的纳米炭;
图11为不同纳米炭的MTT实验结果图,AC-5-Oxi为未经处理的携载Oxi的纳米炭,ACO-30%-Oxi代表仅仅经过氧化处理所得的携载Oxi的纳米炭,ACO-6PEG-Oxi代表本发明的携载Oxi的纳米炭;
图12为本发明纳米炭在不同pH下对5FU的释放结果图;
图13为本发明纳米炭在不同pH下对Oxi的释放结果图。
具体实施方式
下面通过实施例对本发明进行具体描述,有必要在此指出的是以下实施例只是用于对本发明进行进一步的说明,不能理解为对本发明保护范围的限制,该领域的技术熟练人员根据上述发明内容所做出的一些非本质的改进和调整,仍属于本发明的保护范围。
实施例1
(1)原料与试剂
商业活性炭(上海海诺炭业有限公司),常规试剂均购于海鸿实验仪器有限公司;mPEG-NH2(MW 2000)购于苏州诺德派森医药科技有限公司,1-乙基-3-(3-二甲氨丙基)碳二亚胺(EDC,≥99.0%)购于J&K Chemical Ltd.;N-羟基丁二酰亚胺(HOSu,≥98%)购于Alfa Aesar;3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT,商品名:噻唑蓝)购于Sigma;DMEM(Dulbecco’s Modified Eagle Medium)高糖培养基购于HyClone;胎牛血清(FBS)购于HyClone;胰酶购于Gibieo;青霉素-链霉素双抗试剂(100x)购于Gibico;异硫氰酸荧光素异构体(FITC,90%)购于Acros Organics;结肠癌细胞(SW480)购于四川大学华西医学院;上皮细胞(L929)和巨噬细胞(RAW264.7)为本实验室冻存细胞。
(2)活性炭纳米颗粒的制备
制备工艺:
1)将市售活性炭混溶于无水乙醇中(约1g/5mL);
2)将混溶液加入分散砂磨机配套的不锈钢桶中,加入一定量的研磨珠子,在2500rpm转速下打磨;
3)研磨一定时间(分别处理1h,3h和5h)后,停止研磨,先用200目纱布粗过滤,除去珠子,后用500目纱布过滤除去较大粒径的活性炭颗粒;
4)将过滤后的产物用超纯水洗涤数次,120℃烘干,即得到活性炭纳米颗粒(ACNP),分别记为AC-1h、AC-3h和AC-5h,未处理样记为AC-0h。
动态光散射DLS的结果(表1)也近似说明了相同的结果,其数值结果为所有颗粒粒径的统计结算结果,反应了整体粒径尺寸减小,分布变窄的趋势。这一粒径尺寸在淋巴靶向应用的要求范围之内(50 nm<尺寸<500nm),故可以考虑用作淋巴靶向示踪和淋巴靶向制剂的制备,后期如有需要,可进一步对其进行研磨的后续处理,使其符合使用要求,即活性炭颗粒粒径可以通过物理研磨进行初步的调控。本文选用研磨5小时的样品(AC-5h)做后续研究。
表1
(3)氧化活性炭纳米颗粒的制备
制备方法流程:选择粒径参数较为合适的样(AC-5h)进行氧化处理
1)丙酮超声清洗残余有机杂质:按4mL/g的比例配制活性炭纳米颗粒的丙酮混溶液,室温超声处理3h,超声功率为40kHZ(60%);超声处理后抽滤,室温晾干,120℃烘干待进一步处理。
2)水煮沸处理:将烘干的丙酮处理过的超纯水与ACNP以10mL/g的比例加入单颈瓶,磁力搅拌,煮沸(冷凝回流)2h;抽滤,120℃烘干至恒重,样品命名为AC-5。
双氧水氧化处理:称取上一步处理产物活性炭纳米颗粒一定质量于三口瓶中(分别接冷凝回流,恒压滴液漏斗和温度计),先按1mL/g 的比例加入超纯水,再在慢搅拌下,滴加等体积的H2O2氧化剂,反应0.5h;然后升高温度至70℃,逐滴加入4体积(4mL/g)剩余的双氧水,控制3h滴完,滴加完后再充分反应2h;最后进行抽滤,用超纯水洗涤多次至pH呈中性,普通烘箱中先60℃烘3h,再120℃烘干至恒重。配制使用的不同浓度的H2O2氧化剂有6%、15%和30%三个体积浓度,对应氧化产物分别命名为ACO-6%、ACO-15%和ACO-30%。
由表2的Boehmn滴定结果可知,氧化改性后,活性炭表面羧基含量显著的变化,随着氧化剂浓度的提高,羧基含量明显增大,其含量从0.10mmol/g增加到了约0.75mmol/g。本实验中用NaOH中和的样品抽滤较难,活性炭很容易抽滤到下层滤液中,而且滤液颜色也发生了变化,变为暗黄色,最后盐酸滴定终点判断受到干扰,误差可能较大,而NaHCO3中和的滤液为澄清透明液。关于粒径参数更为直观的结果见图1。
表2(单位:mmol/g)
(4)氧化活性炭纳米颗粒接枝mPEG-NH2
采用经典的活化酯缩合反应,将不同投料的mPEG-NH2接枝到表面羧基含量较高的氧化活性炭纳米颗粒(ACO-30%)。接枝工艺如下:称取1g活性炭纳米颗粒于250mL单颈瓶中,加入100mL超纯水,在磁力搅拌下分散1h,再分别加入EDC(1.00mmol)和HOSu(1.00mmol),室温反应1h,最后滴加入mPEG-NH2水溶液,继续常温反应8h。反应结束后,抽滤洗涤滤渣数次,并且将滤渣再用超纯水重新分散,超声振荡,洗去可能吸附的mPEG-NH2,最后将滤渣溶解转移进透析带(截留分子量为3500)中在3L超纯水中透析一周,每隔一定时间换透析用水,充分除去反应试剂和未接枝的mPEG-NH2。透析结束后旋蒸除去大部分水分,后45℃真空干燥24h,密闭保存。样品根据每克活性炭纳米颗粒加入的mPEG-NH2不同(0.1g,0.3g和0.6g)分别命名为ACO-1PEG,ACO-3PEG和ACO-6PEG。(5)氧化活性炭纳米颗粒接枝PEG的表征
傅里叶变换红外光谱(FTIR)
分别取微量接枝PEG的氧化活性炭纳米颗粒,按1%质量比与KBr粉末混合,压片制样,在Nicolet-560型红外吸收光谱分析仪上测定其透射红外谱图,扫描区间为4000~500cm-1,分辨率为4cm-1,检测次数为32次。
热重分析(TGA)
采用Q500型热重分析仪(美国TA仪器公司)在氮气环境下对测试样品进行热重分析,加热范围为50~600℃,升温速率为20℃/min,样品用量范围为5~10mg。
透射电镜(TEM)
取适量改性后活性炭纳米颗粒混溶于1mL无水乙醇中,放置于超声波振荡器中超声振荡20min后用移液器吸取少量混溶液滴于镀碳铜网膜上,待自然晾干,用用Hitachi model H-600-4透射电子显微镜直接观察样品颗粒大小和形态,加速电压为75KV。
激光粒度仪(DLS)
取适量接枝PEG的氧化活性炭纳米颗粒样品,混溶于1mL超纯水中,放置于超声波振荡器中超声振荡20min后,配制稀释成合适浓度后,使用Zetasizer Nano ZS动态光散射(DLS)纳米粒度仪(Malvern,UK)对于其粒径和粒径分布进行测定,测试温度为25℃,散射角为90°。
(6)水分散及稳定性研究
鉴于活性炭混溶液呈黑色,不容易观察其分散及稳定性情况,本文主要关注混溶液的界面(界面:上层较澄清,下层黑浑浊),以此来考察样品的沉降速度,近似表征其溶液稳定性。需在较强光下观察或透过手电筒光观察样品界面。
称取样品1mg于10mL比色管中,然后加入10mL的超纯水,超声2h使各个样品达到完全均匀的分散。然后静置垂直摆放,定时观察记录样品混溶液界面的位置(距离上液面的距离h)。通过计算,可得到静置不同时间后,界面下降高度百分比,即可定性作为样品水溶液稳定性的一个指标。计算公式为:
ν(%)=h/H×100,式中ν表示沉降高度百分比(%),h表示静置一段时间后,混溶液界面离上页面的距离,即下沉距离(单位cm),H表示整个混溶液在比色管中的高度(单位cm),本实验中H=7.40 cm。
待样品完全沉降后,可再考察其在水溶液重新分散的能力,定性的考察主要通过振荡或超声等使其重新达到较好均一分散的难易程度。
(7)接枝PEG的氧化活性炭纳米颗粒对药物的包载
以超纯水为溶剂分别配制一系列浓度含量的5-氟尿嘧啶溶液,待完全溶解后,分别加入改性样品活性炭那纳米颗粒,室温下超声振荡180min。吸附结束后,抽滤取下层滤液,测定其中游离药物的浓度,得到载药量。过滤后的上层载药活性炭纳米颗粒先用超纯水洗涤数次,洗去表面残留的药物。再在普通烘箱60℃初步干燥10min,然后转移进真空干燥24h,温度控制低于45℃。即得到相应的给药制剂,干燥后密闭保存,减少活性炭吸收空气中的水分。
(8)氧化活性炭纳米颗粒接枝PEG给药制剂的释药
分别精密称取一定量的活性炭纳米给药制剂,溶于10mL配好的PB缓冲液中,迅速将溶液转移入预先处理的透析袋中,将透析袋两端扎紧放入预先盛有90mL PB缓冲溶液的广口瓶中。在37℃恒温摇床中进行药物释放,转速为110rpm。定时移取释放液,并补加相同体积的缓冲液。使用HPLC测定释放液中药物的浓度,从而计算得到其释放曲线。
(9)氧化活性炭纳米颗粒接枝PEG的体外细胞实验
细胞培养
常规复苏冻存的巨噬细胞、L929上皮细胞和SW480结肠癌细胞,在加入含10%(v/v)胎牛血清、1%(v/v)双抗(青霉素和链霉素)的高糖DMEM培养基中放于37℃、5%CO2、饱和湿度的培养箱中培养。两种细胞均为贴壁细胞,待其布满培养瓶底部时以胰蛋白酶消化进行传代培养。
细胞内化
将对数生长期细胞进行消化,以每孔1×105个细胞的密度接种到置于6孔板的盖玻片上,于37℃、5%CO2及饱和湿度条件下培养24h至细胞形态良好。吸去孔板中的培养基,分别各自加入1mL FITC标记的活性炭纳米颗粒(FTIC浓度约为50μg/mL)的培养基溶液,继续共培养一定时间。除去培养基,用4%的多聚甲醛溶液于4℃固定30min,再用DAPI于37℃染色10min。最后使用50%甘油溶液封片,并通过CLSM观察。
MTT实验
将对数生长期的细胞消化,以每孔500μL、0.8×104个细胞的密度接种到置于48孔板内中,置于37℃、5%CO2、饱和湿度的培养箱中培养至细胞贴壁且形态良好。去除培养基,每孔加入500μL不同药物浓度的活性炭纳米给药制剂(选用投料比为10%的相应纳米给药制剂作为实验对象),每个浓度设3个复孔,设置空白对照组,于37℃,5%CO2及饱和湿度条件下共培养1d和3d。每孔加入40μL MTT溶液(5mg/mL),继续培养孵育4h。4h后小心吸取孔内培养上清液,按每孔500μL加入DMSO,于摇床低速振荡20min。用0.22μm的有机滤头过滤DMSO溶液,再等量吸取滤液加入96孔板中,在酶标仪(DNM-9602酶标分析仪)492nm波长下测定各孔的吸光度OD值,计算相应的细胞活性(存活率)。
结果与讨论
氧化活性炭纳米颗粒接枝PEG的表征
红外光谱
在红外谱图中(图2),接枝PEG后,在2923cm-1、2854cm-1和1111cm-1等处附近出现了较为明显甲基、亚甲基的对称伸缩振动和醚键的伸缩振动峰,1433cm-1处附件出现碳氢的弯曲振动峰,而且部分峰的吸光度随着PEG投料的增加,有一定程度的增加,定性表示了接枝PEG的成功和接枝量的不同。
热重分析(TGA)
由TGA的结果可知(图3),接枝处理的样品在PEG的分解峰附近处,TG曲线都存在明显的突降过程。对于这一温度范围(300-440℃)各样品的质量变化做近似的估算,得到对应的ΔWeight(表3),由表中结果可知,ACO-3PEG与ACO-6PEG的PEG接枝基本相同,约为8.00%,可能原因是接枝主要发生在活性炭纳米颗粒的表面,表面羧基含量有限,接枝过程达到了饱和状态。两种相比于ACO-1PEG的接枝量有一定程度的提高。
表3
粒径表征
由图4(A)和表4可知,接枝PEG后活性炭粒径未有明显的变化,约在360.0nm左右,较之ACO-30%的粒径结果略有减小,可能由于接枝后亲水性增加,减小了团聚现象,粒径曲线向小粒径尺寸偏移,粒径分布基本不变。图4(B)为ACO-6PEG样品的透射电镜图,可以看出活性炭纳米颗粒的轮廓较为圆滑,接枝PEG后不能在纳米颗粒边缘看出明显的变化,这与活性炭纳米颗粒本身不规则,粒径较大,导致透射电镜检测时颗粒的衬度变化不明显,不易判断有关。
表4
水分散及稳定性研究
样品的沉降速度(高度下降百分比):直观形象的4天沉降结果如图5。活性炭纳米颗粒被较高浓度双氧水氧化后,其沉降速度降低,水稳定性提高。可能是由于较多的极性表面羧基可与水分子发生相互作用,提高了纳米颗粒在水中的分散稳定性。而当氧化活性炭纳米颗粒接枝PEG后,其水稳定性又进一步提高,这可能由于PEG溶于水中,会在颗粒表面溶解伸展形成一定的空间体积排斥,从而增加了活性炭纳米颗粒在水溶液中的稳定性,并且减少了其发生聚沉的可能性。
对完全沉降的样品进行重新分散探究,发现仅氧化改性的样品沉降后会在底部沉积结块,简单摇晃处理并不能有效地使底部的活性炭块团重新分散良好,需要对其进行较长时间的超声处理,如30min。而接枝改性后的样品沉降后在底部不会结成团块,稍微的振荡摇晃即可使底部块团分散开,仅需几分钟的超声就可以重新分散良好。超声处理后,所有样品均表现出良好的复溶分散。透过灯光,ACO-6PEG、ACO-3PEG相比较其它样品而言,分散液颜色略浅,透光性好,其次是氧化系列的样品。
由载药量的结果(图6)可知,相同操作下,接枝PEG后,活性炭纳米颗粒对药物包载能力降低,且随着接枝量的增加,载药量进一步降低。导致载药量降低的原因可能来源于以下几点因素的影响:一是接枝改性后,纳米颗粒中PEG分子对于吸附包载没有什么贡献,且导致药物载体的比表面积的降低;二是PEG分子可能填充了一定的孔体积,而且极有可能堵塞了表面孔洞,导致药物不能有效地进入孔隙,即不能发生吸附相互作用;三是PEG分子的阻碍作用可能导致需要更长的超声吸附时间,让药物跨过阻碍,进入孔隙,达到充分的吸附作用。
由累积释放曲线(图7)可以很明显地看出,ACO-30%接枝PEG后,制备的相应活性炭纳米给药制剂对于药物的释放有明显的减缓效果,缓解了活性炭纳米给药制剂初期的快速释放现象,释放2d后,ACO-6PEG的累积释放量只达到约20%左右,而ACO-30%给药制剂的药物累积释放达到了50%。这种明显的药物缓释来源于PEG在活性炭纳米颗粒表面形成的空间保护或者堵塞载体孔洞,增加了药物释放的难度,释放到环境中需要突破PEG的阻碍作用。这种接枝改性对活性炭纳米颗粒作为药物载体本身的功能缓释性可能造成的影响有待进一步的研究。
氧化活性炭纳米颗粒接枝PEG的体外细胞研究
接枝PEG对纳米颗粒细胞内化的影响
由图8可知,ACO-6PEG样品与巨噬细胞共培养1h后,巨噬细胞对于纳米颗粒仍具有优异的内吞作用。另外,残余的活性炭纳米颗粒更易清洗掉。
SW480结肠癌细胞的CLSM结果(图9)表明,接枝改性后的样品ACO-6PEG,仍然具有肿瘤细胞附着性和很高的细胞内化性能,共培养1h后,细胞就有很显著的细胞染色。
接枝PEG对给药制剂药效的影响
不同给药制剂的MTT结果见图10(5-氟尿嘧啶)和图11(奥沙利铂),A图和B图分别表示1天和3天的药效,接枝改性后样品ACO-6PEG给药制剂的体外抗肿瘤细胞疗效与氧化改性样ACO-30%给药制剂的疗效相比,未有显著变化,共培养3天都有较好的抗肿瘤细胞活性。
- 还没有人留言评论。精彩留言会获得点赞!