一种富马酸二甲酯的制备方法与流程
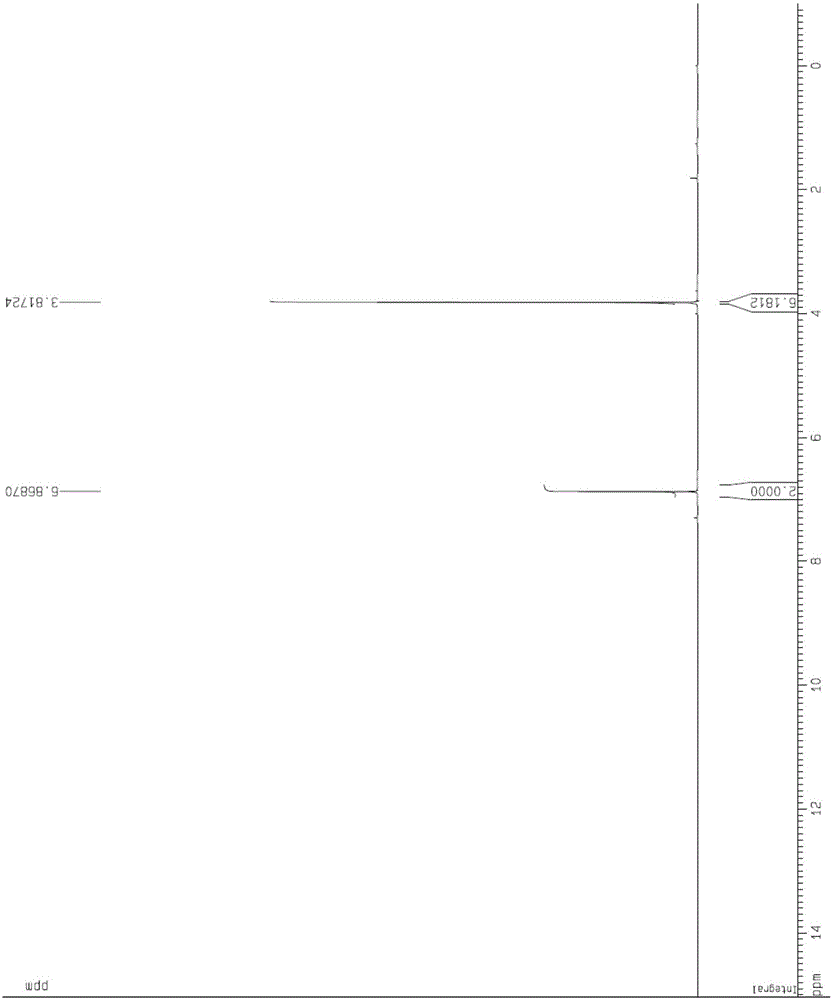
本发明涉及化学领域,特别涉及一种富马酸二甲酯的制备方法。
背景技术:
BG-12(富马酸二甲酯)是一种口服治疗药物,用于治疗成人复发-缓解型多发性硬化症(RRMS),MS中最常见的形式。BG-12由百键艾迪(Biogen Idec)公司制造。成为第三种用于治疗MS的疾病缓解的口服药物,其他两种包括芬戈莫德(Gilenya)和特立氟胺(Aubagio)。相对于现有的MS药物,富马酸二甲酯具有新的作用机制,它被认为可激活核因子样-2转录途径,从而减少导致脱髓鞘的氧化应激反应。
研究机构称,DEFINE及CONFIRM两项安慰剂对照III期临床试验作为批准的基础。这两项试验共纳入2700例患者。除了药物与安慰剂比较,CONFIRM试验还包括第3组----开放标签性醋酸格拉默(Copaxone,一种标准的注射MS药物)组。
随着制剂科学的发展,原料药的晶型和粒度对药物的效果有明显的影响。粒度的大小经常成为影响制剂研发成败的关键因素。
富马酸二甲酯原料药的粒度控制方法为物理粉碎和重结晶。一般物理粉碎过程都会来带来大量的粉尘,造成粉尘污染;富马酸二甲酯难溶于水,粉碎机的清洁过程繁琐;富马酸二甲酯对皮肤有严重的刺激作用,粉碎过程中产生的大量粉尘极易吸附在人的身体上造成皮肤过敏,给生产人员带来伤害。重结晶方法是一种常用的控制原料药粒度和晶型的手段。专利WO2012170923中关于富马酸二甲酯的粒度和晶型控制中提到采用各种有机溶剂、水或混合溶剂通过重结晶可以得到具有一定晶型的富马酸二甲酯,然后采用气流粉碎机(jet milling)粉碎可以得到具有一定粒度范围的富马酸二甲酯;专利中指出通过粉碎粒度可以控制在20-250微米之间。该专利中控制富马酸二甲酯的晶型和粒度经两个操作过程进行控制——分别为重结晶控制和粉碎控制,步 骤繁琐;粉碎过程中富马酸二甲酯粉尘对人体有严重的刺激性反应,对操作人员可能带来伤害,在放大生产中对工人存在较大的的安全风险。
技术实现要素:
有鉴于此,本发明提供一种富马酸二甲酯的制备方法。该方法采用化学手段一步法控制BG-12原料药晶型和粒度的方法。该方法只需一个操作过程即可实现现有技术中重结晶和粉碎两个操作过程,得到符合制药生产要求的粒度,避免给操作者带来严重的刺激性反应,大大提高了生产安全性。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种富马酸二甲酯的制备方法,包括如下步骤:
步骤1:取富马酸、硫酸和甲醇经过酯化反应,制备获得富马酸二甲酯粗品;
步骤2:取步骤1获得的富马酸二甲酯粗品经有机溶剂溶解,碱性溶液洗涤,获得富马酸二甲酯溶液;
步骤3:取步骤2获得的富马酸二甲酯溶液除去溶剂,与重结晶溶剂混合后重结晶,获得富马酸二甲酯;
步骤2中的所述碱性溶液包括K2CO3、Na2CO3、KHCO3、NaHCO3、NaOH、KOH;
步骤3中的所述重结晶溶剂选自MeOH、EtOH、THF、Acetone、CH3CN、i-PrOH、DMF、NMP、DMSO和水。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法制得的所述富马酸二甲酯粒度的d(0.5)为10~35μm、50~75μm、80~105μm或110~130μm。
在本发明的一些具体实施方案中,所述富马酸二甲酯粒度d(0.5)10-35μm的工艺参数:转速200~250转/min,降温速率2~5min/℃,溶剂与水的体积比1:3~5;
所述富马酸二甲酯粒度d(0.5)50-75μm工艺参数:转速200~250转/min,降温速率2~3min/℃,溶剂与水的体积比1:2~4;
所述富马酸二甲酯粒度d(0.5)80-105μm工艺参数:转速180~250转/min,降温速率3~4min/℃,溶剂与水的体积比1:0.5~2;
所述富马酸二甲酯粒度d(0.5)80-105μm工艺参数:转速150~175转/min,降温速率4~5min/℃,溶剂与水的体积比4~5:1;
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中所述重结晶的搅拌速度为150~250转/min。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中所述重结晶的降温速率为1~5min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中重结晶溶剂为MeOH和水,其中MeOH和水的体积比为5:1~1:5。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,以kg/L计,所述富马酸二甲酯与所述重结晶溶剂的质量体积比为1:2~1:5。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为5:1:3~5,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为10~35μm;优选25.5μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为1:3~5,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为10~35μm;优选25.5μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为乙腈和水,乙腈和水的体积比为1:1~2,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选89.7μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为四氢呋喃和水,乙腈和水的体积比为1:4~5,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为13.6μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为四氢呋喃和水,乙腈和水的体积比为25~28:125,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为13.6μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为4~5:1,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为4~5min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为110~130μm;优选118.9μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为46~47:9,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为4~5min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为110~130μm;优选118.9μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为丙酮和水,丙酮和水的体积比为1~2:1,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选93.1μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为丙酮和水,丙酮和水的体积比为11~12:6,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选93.1μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为异丙醇和水,异丙醇和水的体积比为1:2~3,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为50~75μm;优选63.7μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为异丙醇和水,异丙醇和水的体积比为25~26:50,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为50~75μm;优选63.7μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为DMF和水,异丙醇和水的体积比为1~2:1,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为4~5min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选85.0μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为DMF和水,异丙醇和水的体积比为8~9:9,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为4~5min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选85.0μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为1~2:1,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选90.0μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为20~21:20,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为2~3min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为80~105μm;优选90.0μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为1:4~5,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为1~2min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为13.5μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,所述重结晶溶剂为MeOH和水,MeOH和水的体积比为22~23:110,所述重结晶的搅拌速度为150~250转/min,所述重结晶的降温速率为1~2min/℃,温度回至15~35℃后继续搅拌过夜、离心、减压干燥,获得所述富马酸二甲酯的粒度为13.5μm。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,步骤2中所述有机溶剂包括EtOAc、CH2Cl2、CHCl3、Et2O、MTBE,优选CH2Cl2。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,步骤2中的所述碱性溶液包括NaOH、KHCO3或NaHCO3。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,步骤3中的所述重结晶溶剂中的水的温度为0℃。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,富马酸二甲酯粗品的合成方法为:于反应釜中加入甲醇,慢慢加入硫酸,搅拌速度约150±50转/min;待硫酸甲醇溶液配制完成后,加入富马酸,加热回流反应8小时以上,HPLC检测原料反应完全即可停止反应。停止反应后将反应趁热泵入盛有140L冰水的容器中,操作过程中不断搅拌,随着温度的降低大量白色粉末固体析出,继续搅拌1小时,采用400目的滤布离心即可得到富马酸二甲酯粗品。其中富马酸、硫酸和甲醇的质量体积比为:2kg:0.2kg:10L。
更具体为:于100L反应釜中加入70L甲醇,慢慢加入硫酸1.4kg,搅拌速度约150±50转/min;待硫酸甲醇溶液配制完成后,加入富马酸14kg(120mol),加热回流反应8小时以上,HPLC检测原料反应完全即可停止反应。停止反应后将反应趁热泵入盛有140L冰水的容器中,操作过程中不断搅拌,随着温度的降低大量白色粉末固体析出,继续搅拌1小时,采用400目的滤布离心即可得到富马酸二甲酯粗品。
在本发明的一些具体实施方案中,富马酸二甲酯的制备方法中,富马酸二甲酯粗品的后处理——制备富马酸二甲酯溶液的方法具体为:将富马酸二甲酯粗品用有机溶剂溶解后,加入碱性溶液,搅拌10分钟,转速150±50转/min,停止搅拌后静置分层,分出下层水相,上层有机相继续使用碱性溶液洗涤1遍,洗涤溶液用量30L/次,保证水相溶液pH为7。有机相再用5kg无水硫酸钠干燥30分钟,过滤分离得到富马酸二甲酯的乙酸乙酯溶液。
更具体为:将富马酸二甲酯粗品用84L乙酸乙酯溶解后加入到150L的萃取釜中,加入饱和的碳酸氢钠溶液30L,搅拌10分钟,转速150±50转/min,停止搅拌后静置分层,分出下层水相,上层有机相继续使用饱和的碳酸氢钠洗涤两遍、饱和氯化钠溶液洗涤1遍,洗涤溶液用量30L/次,保证水相溶液pH为7。有机相再用5kg无水硫酸钠干燥30分钟,过滤分离得到富马酸二甲酯的乙酸乙酯溶液。
本发明采用水溶性的有机溶剂进行溶解,然后利用一定体积的水进行重结晶即可。整个结晶过程中析晶速度快(一般在30分钟即可完成操作),富马酸二甲酯的粒度和晶型均可以达到制剂要求。通过采用水溶性溶剂和水混合结晶的方法即可达到WO2012170923中提到的繁琐实验过程。因此本发明具有明显的创新性,操作过程简单、可控,实验结果稳定、整个过程可以在密闭的反应釜中进行,能够最大程度的避免人员伤害。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
图1示富马酸二甲酯的合成工艺流程图;
图2示实施例3制得的富马酸二甲酯的氢谱图;
图3示实施例3制得的富马酸二甲酯的碳谱图;
图4示实施例3制得的富马酸二甲酯的GC-MS谱图;
图5示实施例3制得的富马酸二甲酯的粒度分布图;
图6示实施例3制得的富马酸二甲酯的X射线粉末衍射图;其中图6(A)示X射线粉末衍射图谱;图6(B)示X射线粉末衍射数据;
图7示实施例6制得的富马酸二甲酯的粒度分布图;
图8示实施例6制得的富马酸二甲酯的X射线粉末衍射图;其中图8(A)示X射线粉末衍射图谱;图8(B)示X射线粉末衍射数据;
图9示实施例9制得的富马酸二甲酯的粒度分布图;
图10示实施例10制得的富马酸二甲酯的粒度分布图;
图11示实施例11制得的富马酸二甲酯的粒度分布图;
图12示实施例12制得的富马酸二甲酯的粒度分布图;
图13示实施例13制得的富马酸二甲酯的粒度分布图;
图14示实施例14制得的富马酸二甲酯的粒度分布图;
图15示实施例15制得的富马酸二甲酯的粒度分布图。
具体实施方式
本发明公开了一种富马酸二甲酯的制备方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明的内容为富马酸二甲酯的后处理工艺、晶型和粒度的控制工艺,通过工艺的改进可以很好的避免富马酸粉尘污染和皮肤刺激危害。具体工艺流程如图1所示:
一方面富马酸二甲酯难溶于水,所以它的粒度和晶型是制剂工艺中需要控制的关键参数;另一方面由于富马酸二甲酯具有强烈的皮肤刺激性,采用简便易操作的化学方法控制晶型和粒度十分必要。本发明工艺具有操作简便、安全、便于放大;产物收率、纯度高等特点。本工艺具有强大的市场竞争力和非常可观的经济价值,根本上解决了富马酸二甲酯粉碎过程中带来的刺激性反应。该方法在富马酸二甲酯粒度和晶型控制上具有明显的优势。
合成工艺路线如下:
本发明提供的富马酸二甲酯的制备方法中所用原料及试剂均可由市场购得。
说明书和权利要求书中所使用的缩写的含义如下:
THF 四氢呋喃
DMF N,N-二甲基甲酰胺
METB 甲基叔丁基醚
DMSO 二甲基亚砜
NaOH 氢氧化钠
KOH 氢氧化钾
NaHCO3 碳酸氢钠
KHCO3 碳酸氢钾
MeOH 甲醇
EtOH 乙醇
i-PrOH 异丙醇
CH3CN 乙腈
Actone 丙酮
EtOAc 乙酸乙酯
NMP N-甲基吡咯烷酮
CH2Cl2 二氯甲烷
CHCl3 三氯甲烷
Et2O 乙醚
下面结合实施例,进一步阐述本发明:
实施例1:富马酸二甲酯粗品的合成
于100L反应釜中加入70L甲醇,慢慢加入硫酸1.4kg,搅拌速度约150±50转/min;待硫酸甲醇溶液配制完成后,加入富马酸14kg(mol),加热回流反应8小时以上,HPLC检测原料反应完全即可停止反应。停止反应后将反应趁热泵入盛有140L冰水的容器中,操作过程中不断搅拌,随着温度的降低大量白色粉末固体析出,继续搅拌1小时,采用400目的滤布离心即可得到富马酸二甲酯粗品。粗品有待进一步纯化。
富马酸二甲酯粗品的合成
实施例2:富马酸二甲酯粗品的后处理
实施例1中得到的富马酸二甲酯粗品用84L乙酸乙酯溶解后加入到150L的萃取釜中,加入饱和的碳酸氢钠溶液30L,搅拌10分钟,转速150±50转/min,停止搅拌后静置分层,分出下层水相,上层有机相继续使用饱和的碳酸氢钠洗涤两遍、饱和氯化钠溶液洗涤1遍,洗涤溶液用量30L/次,保证水相溶液pH为7。有机相再用5kg无水硫酸钠干燥30分钟,过滤分离得到富马酸二甲酯的乙酸乙酯溶液。富马酸二甲酯粗品的合成具体流程如下所示:
实施例3:富马酸二甲酯的粒度和晶型控制过程
将实施例2得到的富马酸二甲酯的乙酸乙酯溶液加入到蒸馏釜中,减压蒸馏除去溶剂(减压蒸馏过程中溶液温度小于50℃),采用甲醇蒸馏替换反应釜中的乙酸乙酯溶剂,通过多次蒸馏替换,当反应釜中乙酸乙酯的含量小于5%时进行富马酸二甲酯的粒度和晶型控制实验。补加甲醇溶液保持溶液体积在45~50L,继续升温至溶液回流,当固体全部溶解后将反应液泵入装有约150L冰水的反应釜中,混合过程中伴随搅拌,搅拌速度200±50转/min,降温速率2min/℃、温度回至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯14.5kg,收率83%。产品采用GC-MS、NMR进行 鉴定,采用马尔文粒度仪和锐影(Empyrean)X射线衍射仪进行粒度和晶型表征,粒度测试d(0.5)=25.5微米。
1H-NMR(CDCl3)δ:6.87(s,2H),3.82(s,6H);
13C-NMR(CDCl3)δ:165.3,133.3,52.2ppm。
碎片离子峰113:
碎片离子峰85和59:
通过核磁和质谱确认产物为富马酸二甲酯。
粒度分布测定检测条件:干法测定;
样品为细粉,遮光度:3.86%,残差:0.38%,分散器气压0.4MPa。
X射线粉末衍射测定:测定方法JY/T009-1996转靶多晶体X射线衍射方法通则
实施例4:富马酸二甲酯粗品的合成
于20L反应釜中加入15L甲醇,慢慢加入硫酸0.3kg,搅拌速度约150±50转/min;待硫酸甲醇溶液配制完成后,加入富马酸3kg(mol),加热回流反应8小时以上,HPLC检测原料反应完全即可停止反应。停止反应后将反应趁热泵入盛有45L冰水的反应釜中,操作过程中不断搅拌,随着温度的降低大量白色粉末固体析出,继续搅拌1小时,采用400目的滤布离心即可得到富马酸二甲酯粗品。
实施例5:富马酸二甲酯粗品的后处理
实施例4中得到的富马酸二甲酯粗品用15L二氯甲烷溶解后加入到20L的萃取釜中,加入饱和的碳酸氢钾溶液2L,搅拌10分钟,转速150±50转/min,停止搅拌后静置分层,分出下层层有机相,上层水相继续使用饱和的碳酸氢钾洗涤两遍、饱和氯化钠溶液洗涤1遍,洗涤溶液用量2L/次,保证水相溶液pH为7。下层有机相再用0.5kg无水硫酸钠干燥30分钟,过滤分离得到富马酸二甲酯的二氯甲烷溶液。
实施例6:富马酸二甲酯的粒度和晶型控制过程
将实施例5得到的富马酸二甲酯的二氯甲烷溶液加入到蒸馏釜中,减压蒸馏除去溶剂(减压蒸馏过程中溶液温度小于50℃),采用乙腈蒸馏替换反应釜中的二氯甲烷溶剂,通过多次蒸馏替换,当反应釜中二氯甲烷的含量小于5%时进行富马酸二甲酯的粒度和晶型控制实验。补加乙腈溶持溶液体积在9-10L,继续升温至溶液回流,当固体全部溶解后停止加热、向反应釜中泵入9L冰水,混合过程中伴随搅拌,搅拌速度200±50转/min,降温速度3min/℃、温度回至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯3.3kg,收率88%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,d(0.5)=89.7微米。
实施例7:富马酸二甲酯粗品的合成
于100L反应釜中加入40L甲醇,慢慢加入硫酸0.7kg,搅拌速度约150±50转/min;待硫酸甲醇溶液配制完成后,加入富马酸7kg,加热回流反应8小时以上,HPLC检测原料反应完全即可停止反应。反应停止后停止加热,改用冷却液冷却,向反应釜中加入60L冰水,剧烈搅拌,当釜内温度降低至20℃后继续搅拌2小时,采用400目的滤布离心即可得到富马酸二甲酯粗品。
实施例8:富马酸二甲酯粗品的后处理
实施例7中得到的富马酸二甲酯粗品用56L甲基叔丁基醚溶解后加入到100L的萃取釜中,加入1%的氢氧化钠溶液14L,搅拌10分钟,转速150±50转/min,停止搅拌后静置分层,分出下层层水相,上层有机相继续使用1%氢 氧化钠溶液洗涤两遍、饱和氯化钠溶液洗涤1遍,洗涤溶液用量14L/次,保证水相溶液pH为7。上层有机相再用2kg无水硫酸钠干燥30分钟,过滤分离得到富马酸二甲酯的甲基叔丁基醚溶液。
实施例9:富马酸二甲酯的粒度和晶型控制过程
将实施例8得到的富马酸二甲酯的甲基叔丁基醚溶液加入到蒸馏釜中,减压蒸馏除去溶剂(减压蒸馏过程中溶液温度小于50℃),采用四氢呋喃蒸馏替换反应釜中的甲基叔丁基醚溶剂,通过多次蒸馏替换,当反应釜中甲基叔丁基醚的含量小于5%时进行富马酸二甲酯的粒度和晶型控制实验。补加四氢呋喃,保持溶液体积在25-26L,继续升温至溶液回流,当固体全部溶解后停止加热、采用冷媒冷却溶液同时向釜中加入125L冰水,溶液混合过程中剧烈搅拌,搅拌速度200±50转/min,降温速率2min/℃、溶液温度回至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯6.9kg,收率79%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,晶型与实施例3和6相同,粒度d(0.5)=13.6微米。
实施例10:富马酸二甲酯的粒度和晶型控制过程
将15.3kg富马酸二甲酯加入到46-47L甲醇中,加热溶解,当富马酸二甲酯全部溶解后,停止加热采用冷媒冷却溶液同时向釜中加入9L冰水,混合过程中伴随搅拌,搅拌速度200±50转/min,降温速率4min/℃、温度回至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯13.8kg,收率90%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,晶型与实施例3和6相同粒度d(0.5)=118.9微米。
实施例11:富马酸二甲酯的粒度和晶型控制过程
将4.4kg富马酸二甲酯加入到11-12L丙酮中,加热溶解,当富马酸二甲酯全部溶解后,停止加热,然后将热的富马酸二甲酯丙酮溶液迅速倒入装有6L冰水的反应釜中,混合过程中快速搅拌,搅拌速度200±50转/min,溶液混合完毕后,控制降温速率3min/℃、温度回至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯4.1kg,收率92%。采用马尔文 粒度仪和X-粉末衍射仪进行粒度和晶型表征,晶型与实施例3和6相同,粒度d(0.5)=93.1微米。
实施例12:富马酸二甲酯的粒度和晶型控制过程
将5.1kg富马酸二甲酯加入到25-26L异丙醇中,加热溶解,当富马酸二甲酯全部溶解后,停止加热采用冷媒冷却溶液同时向釜中加入50L冰水,混合过程中伴随搅拌,搅拌速度200±50转/min,降温速率2min/℃、降至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯4.5kg,收率88%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,晶型与实施例3和6相同,粒度d(0.5)=63.7微米。
实施例13:富马酸二甲酯的粒度和晶型控制过程
将4.8kg富马酸二甲酯加入到8-9L DMF中,加热溶解,当富马酸二甲酯全部溶解后,停止加热采用冷媒冷却溶液同时向釜中加入9L冰水,混合过程中伴随搅拌,搅拌速度200±50转/min,降温速率5min/℃、降至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯4.0kg,收率84%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,晶型与实施例3和6相同,粒度d(0.5)=85.0微米。
实施例14:富马酸二甲酯的粒度和晶型控制过程
将4.2kg富马酸二甲酯加入到20-21L甲醇中,加热溶解,当富马酸二甲酯全部溶解后,停止加热采用冷媒冷却溶液同时向釜中加入20L冰水,混合过程中伴随搅拌,搅拌速度200±50转/min,降温速率2min/℃、降至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯3.6kg,收率86%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,粒度d(0.5)=90.0微米。
实施例15:富马酸二甲酯的粒度和晶型控制过程
将5.6kg富马酸二甲酯加入到22-23L甲醇中,加热溶解,当富马酸二甲酯全部溶解后,迅速将热的富马酸二甲酯溶液泵入110L冰水,混合过程中伴 随剧烈搅拌,搅拌速度200±50转/min,降温速率2min/℃、溶液温度降至室温后继续搅拌过夜、离心、减压干燥得具有一定粒度和晶型的富马酸二甲酯5.1kg,收率91%。采用马尔文粒度仪和X-粉末衍射仪进行粒度和晶型表征,粒度d(0.5)=13.5微米。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!