一种3,3—联二硫酸乙烯酯的生产方法与流程
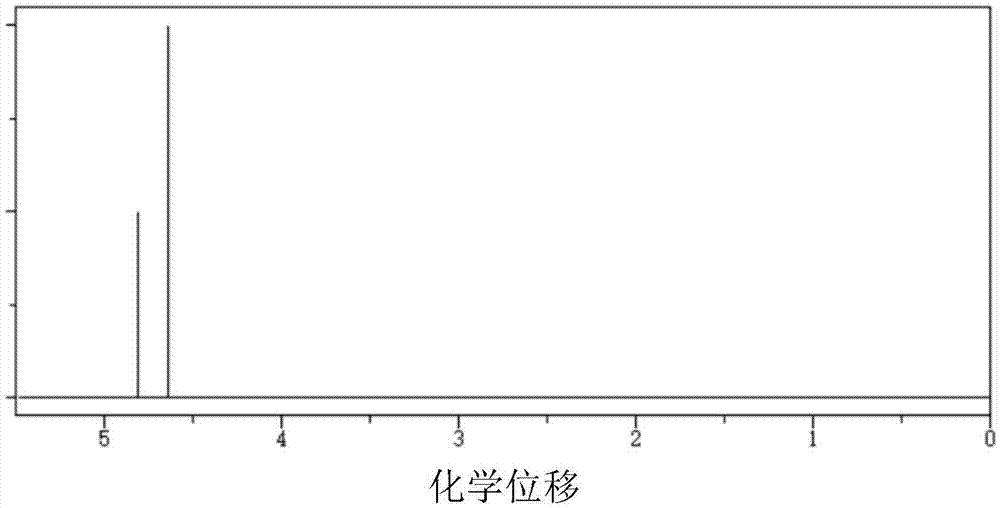
本发明属于化学电源的
技术领域:
,具体涉及一种生产3,3-联二硫酸乙烯酯的方法。
背景技术:
:随着我国的电子信息产业的发展,对于化学电源的需求量越来越大,对其性能要求越来越高。由于锂离子电池具有体积小、安全性能好、重量轻、比能量高、电压高、寿命长、无污染等其它化学电源所无法比拟的优点,目前它已经成为手机、掌上电脑、笔记本电脑、微型摄像机数码照相机等便携式电子设备的主要电源。近年来,锂离子电池的基础研究和应用开发成为热点之一。锂电池中包括正极、负极、电解液和隔膜,但是,电池在充放电过程中,会释放热量,造成电池性能降低。为避免电池性能的降低,往往在电解液中使用添加剂,用于提高高温情况下的电池性能。在制备添加剂时,采用四氯化碳作为溶剂,用于提供极性,在反应过程中会生成氯化氢并使其溶解在四氯化碳内,对反应的进程有影响并对后期溶剂的处理麻烦,不易清除,而且四氯化碳是有毒产品,对人体有害并建议不予以使用;同时在反应中使用高碘酸钠和三氯化钌,使得通过氧化生产的3,3-联二硫酸乙烯酯易产生水解,收率非常低,而且纯度差,进而影响其在电池中的表现性能。技术实现要素:本发明的目的在于客服现有技术的不足,提供一种生产3,3-联二硫酸乙烯酯的方法,旨在避免四氯化碳的使用,同时能够确保目标产物3,3-联二硫酸乙烯酯的使用性能。本发明的技术目的通过下述技术方案予以实现:一种3,3—联二硫酸乙烯酯的生产方法,按照下述步骤进行:步骤1,以氯化亚砜和赤藓糖醇作为反应原料进行加成反应,以得到中间体3,3-联二亚硫酸乙烯酯;将氯化亚砜加入到第一反应釜中并降温至-15—-5℃,滴加赤藓糖醇,在-15—-5℃条件下充分反应,以得到第一反应液,再将第一反应液进行减压蒸馏,蒸出中间体3,3- 联二亚硫酸乙烯酯并通过冷凝回流方式回流到第二反应釜中;在步骤1中,第一反应釜中的温度为-15—-10℃。在步骤1中,氯化亚砜和赤藓糖醇的摩尔比为(1—3):1,优选(1.5—2):1。在步骤1中,采用匀速滴加方式向第一反应釜中滴加赤藓糖醇,用时至少1小时,优选1-2h。在步骤1中,第一反应釜中的反应时间至少为1小时,优选1—3小时,反应时间包括滴加赤藓糖醇的用时,或者在滴加赤藓糖醇之后继续进行反应的时间。在步骤1中,在第一反应釜中进行机械搅拌或者超声分散,以使氯化亚砜和赤藓糖醇充分接触和反应。步骤2,中间体氧化反应得到3,3-联二硫酸乙烯酯;向第二反应釜中加入二氯甲烷和碳酸氢钠并降温至5—10℃,再向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,在5—10℃条件下充分反应,得到水相和有机相共存的第二反应液,静置分层后将水相分离出去,得到的有机相即为3,3-联二硫酸乙烯酯粗产品;在步骤2中,第二反应釜中的温度为5—8℃。在步骤2中,加入第二反应釜的二氯甲烷的用量,以体积计,为步骤1所得中间体3,3-联二亚硫酸乙烯酯体积的1—10倍。在步骤2中,向第二反应釜中加入碳酸氢钠,以使pH值为7-9。在步骤2中,在次氯酸钠和催化剂的水相混合液中,催化剂为三氯化钌或二氧化钌,次氯酸钠和催化剂的摩尔比为1:(0.000001-0.01),次氯酸钠与步骤1所得中间体的摩尔比为(1-6):1。在步骤2中,在第二反应釜中进行机械搅拌或者超声分散,以使各个组份充分接触和反应。在步骤2中,采用匀速滴加方式向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,用时至少1小时,优选1-3h。在步骤2中,第二反应釜中的反应时间至少为1小时,优选1—3小时,反应时间为滴加次氯酸钠和催化剂的水相混合液完毕后的保温时间。在步骤2中,水相和有机相共存的第二反应液静置分层进行分液处理,得到水相和有机相,所述的水相用二氯甲烷萃取三次,得到二氯甲烷萃取液,二氯甲烷萃取液与分 液处理得到的有机相合并,然后将合并后的有机相常压蒸馏出二氯甲烷,得到3,3-联二硫酸乙烯酯粗产品。步骤3,将步骤2得到的3,3-联二硫酸乙烯酯粗产品进行蒸馏,以得到精制的3,3-联二硫酸乙烯酯;在步骤3中,选择在60—80℃条件下对3,3-联二硫酸乙烯酯粗产品进行分子蒸馏,优选70—80摄氏度。本发明技术方案采用氯化亚砜和赤藓糖醇为原料,制备中间体3,3-联二亚硫酸乙烯酯,再利用氧化反应得到3,3-联二硫酸乙烯酯,如附图3所示。以核磁共振对中间体和最终产物进行表征,如附图1和2所示,中间体3,3-联二亚硫酸乙烯酯:1H-NMR(400MHZ,CD3CN)δ(PPM):3.68(S,4H),5.01(S,2H);最终产物3,3-联二硫酸乙烯酯:1H-NMR(400MHZ,CD3CN)δ(PPM):4.53(S,4H),4.84(S,2H)。以最终产物进行产品气相图谱测试,与核磁共振基本表现相同的最终产品信息。色谱柱型号:RTX-65,30m×0.32mm×0.25μm;检测条件:进样口温度:200℃,分流比:100:1;柱流速:1ml/min,柱箱温度(程序升温):初温40℃,保持5min,一阶升温速率20℃/min,二阶温度220℃,保持5min;检测器:FID检测器,温度:290℃,氢气流量:30ml/min,空气流量:400ml/min,尾吹流量:25ml/min,恒定柱流+尾吹:26ml/min;样品制备:1g样品溶于9g溶剂中,进样量:0.5μl。删除溶样溶剂峰及溶样溶剂杂质峰后,面积归一化含量即为产品纯度,产品中残留溶剂的面积归一化含量即为溶剂残留。保留时间峰高峰面积峰面积%17.8931131826898810.0576318.74719304811307260.0944619.0251324558568780.0715921.033157456961119431533799.77631总数1578956461196992822100.00000本发明在加成反应的过程中没有加入任何的溶剂,通过控制加成反应的反应条件,同样可以为反应提供极性,促成加成反应的进行,生成的中间体纯度收率都大大提高,为后期制备的3,3-联二硫酸乙烯酯的纯度和收率提供保障;在氧化反应的过程中加入了二氯甲烷作为溶剂,使生成的3,3-联二硫酸乙烯酯溶于二氯甲烷中,避免了其发生水解,大大提高了3,3-联二硫酸乙烯酯收率,pH值为碱性状态6-9;最后通过(分子)蒸馏的方法,提高3,3-联二硫酸乙烯酯的纯度,并使最终产品的水分含量低。在3,3-联二硫酸乙烯酯使用的电池领域,电池对于水分还有酸值要求很高,水分高,电池性能下降,酸值高,电池寿命低,利用本发明技术方案制备的3,3-联二硫酸乙烯酯纯度可达99.5%以上,水分含量≤20PPM,酸值≤50PPM,用作电池的电解液添加剂之后在负极形成SEI膜,有效的保护负极,不仅防止高温下容量的降低,还可有效提高高温情况下电池的性能。附图说明图1是本发明技术方案中制备的中间体3,3-联二亚硫酸乙烯酯的核磁共振谱图。图2是本发明技术方案制备的3,3-联二硫酸乙烯酯的核磁共振谱图。图3是本发明技术方案的合成路线示意图。图4是添加利用本发明技术方案制备的3,3-联二硫酸乙烯酯作为电池添加剂和电池,与不进行添加的电池的循环性能对比图,其中菱形为不进行添加的空白组,方形为进行添加的实验组。具体实施方式下面结合具体实施例进一步说明本发明的技术方案。实施例1步骤1,以氯化亚砜和赤藓糖醇作为反应原料进行加成反应,以得到中间体3,3-联二亚硫酸乙烯酯;将氯化亚砜加入到第一反应釜中并降温至-15℃,滴加赤藓糖醇,在-15℃条件下充分反应,以得到第一反应液,再将第一反应液进行减压蒸馏,蒸出中间体3,3-联二亚硫酸乙烯酯并通过冷凝回流方式回流到第二反应釜中;氯化亚砜和赤藓糖醇的摩尔比为3:1;采用匀速滴加方式向第一反应釜中滴加赤藓糖醇,用时1小时;反应时间为1小时,在滴加赤藓糖醇之后继续进行反应的时间;在第一反应釜中进行机械搅拌,以使氯化亚砜和赤藓糖醇充分接触和反应。步骤2,中间体氧化反应得到3,3-联二硫酸乙烯酯;向第二反应釜中加入二氯甲烷和碳酸氢钠并降温至5℃,再向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,在5℃条件下充分反应,得到水相和有机相共存的第二反应液,静置分层后将水相分离出去,得到的有机相即为3,3-联二硫酸乙烯酯粗产品;加入第二反应釜的二氯甲烷的用量,以体积计,为步骤1所得中间体3,3-联二亚硫酸乙烯酯 体积的10倍;向第二反应釜中加入碳酸氢钠,以使pH值为9;在次氯酸钠和催化剂的水相混合液中,催化剂为三氯化钌,次氯酸钠和催化剂的摩尔比为1:0.01,次氯酸钠与步骤1所得中间体的摩尔比为1:1,采用匀速滴加方式向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,用时1小时;第二反应釜中的反应时间为1小时;在第二反应釜中进行机械搅拌,以使各个组份充分接触和反应;水相和有机相共存的第二反应液静置分层进行分液处理,得到水相和有机相,所述的水相用二氯甲烷萃取三次,得到二氯甲烷萃取液,二氯甲烷萃取液与分液处理得到的有机相合并,然后将合并后的有机相常压蒸馏出二氯甲烷,得到3,3-联二硫酸乙烯酯粗产品。步骤3,将步骤2得到的3,3-联二硫酸乙烯酯粗产品进行蒸馏,以得到精制的3,3-联二硫酸乙烯酯;选择在80℃条件下对3,3-联二硫酸乙烯酯粗产品进行分子蒸馏。实施例2步骤1,以氯化亚砜和赤藓糖醇作为反应原料进行加成反应,以得到中间体3,3-联二亚硫酸乙烯酯;将氯化亚砜加入到第一反应釜中并降温至-10℃,滴加赤藓糖醇,在-10℃条件下充分反应,以得到第一反应液,再将第一反应液进行减压蒸馏,蒸出中间体3,3-联二亚硫酸乙烯酯并通过冷凝回流方式回流到第二反应釜中;氯化亚砜和赤藓糖醇的摩尔比为2:1;采用匀速滴加方式向第一反应釜中滴加赤藓糖醇,用时2小时;反应时间为3小时,反应时间包括滴加赤藓糖醇的用时,和在滴加赤藓糖醇之后继续进行反应的时间;在第一反应釜中进行超声分散,以使氯化亚砜和赤藓糖醇充分接触和反应。步骤2,中间体氧化反应得到3,3-联二硫酸乙烯酯向第二反应釜中加入二氯甲烷和碳酸氢钠并降温至10℃,再向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,在10℃条件下充分反应,得到水相和有机相共存的第二反应液,静置分层后将水相分离出去,得到的有机相即为3,3-联二硫酸乙烯酯粗产品;加入第二反应釜的二氯甲烷的用量,以体积计,为步骤1所得中间体3,3-联二亚硫酸乙烯酯体积的1倍;向第二反应釜中加入碳酸氢钠,以使pH值为8;在次氯酸钠和催化剂的水相混合液中,催化剂为三氯化钌,次氯酸钠和催化剂的摩尔比为1:0.000001,次氯酸钠与步骤1所得中间体的摩尔比为6:1,采用匀速滴加方式向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,用时3小时;第二反应釜中的反应时间为3小时;在第二反应釜中进行超声分散,以使各个组份充分接触和反应;水相和有机相共存的第二反应 液静置分层进行分液处理,得到水相和有机相,所述的水相用二氯甲烷萃取三次,得到二氯甲烷萃取液,二氯甲烷萃取液与分液处理得到的有机相合并,然后将合并后的有机相常压蒸馏出二氯甲烷,得到3,3-联二硫酸乙烯酯粗产品。步骤3,将步骤2得到的3,3-联二硫酸乙烯酯粗产品进行蒸馏,以得到精制的3,3-联二硫酸乙烯酯;选择在70℃条件下对3,3-联二硫酸乙烯酯粗产品进行分子蒸馏。实施例3步骤1,以氯化亚砜和赤藓糖醇作为反应原料进行加成反应,以得到中间体3,3-联二亚硫酸乙烯酯;将氯化亚砜加入到第一反应釜中并降温至-5℃,滴加赤藓糖醇,在-5℃条件下充分反应,以得到第一反应液,再将第一反应液进行减压蒸馏,蒸出中间体3,3-联二亚硫酸乙烯酯并通过冷凝回流方式回流到第二反应釜中;氯化亚砜和赤藓糖醇的摩尔比为1:1;采用匀速滴加方式向第一反应釜中滴加赤藓糖醇,用时1.5小时;反应时间为3小时,反应时间为在滴加赤藓糖醇之后继续进行反应的时间;在第一反应釜中进行超声分散,以使氯化亚砜和赤藓糖醇充分接触和反应。步骤2,中间体氧化反应得到3,3-联二硫酸乙烯酯;向第二反应釜中加入二氯甲烷和碳酸氢钠并降温至8℃,再向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,在8℃条件下充分反应,得到水相和有机相共存的第二反应液,静置分层后将水相分离出去,得到的有机相即为3,3-联二硫酸乙烯酯粗产品;加入第二反应釜的二氯甲烷的用量,以体积计,为步骤1所得中间体3,3-联二亚硫酸乙烯酯体积的5倍;向第二反应釜中加入碳酸氢钠,以使pH值为7;在次氯酸钠和催化剂的水相混合液中,催化剂为二氯化钌,次氯酸钠和催化剂的摩尔比为1:0.01,次氯酸钠与步骤1所得中间体的摩尔比为3:1,采用匀速滴加方式向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,用时2小时;第二反应釜中的反应时间为2小时;在第二反应釜中进行超声分散,以使各个组份充分接触和反应;水相和有机相共存的第二反应液静置分层进行分液处理,得到水相和有机相,所述的水相用二氯甲烷萃取三次,得到二氯甲烷萃取液,二氯甲烷萃取液与分液处理得到的有机相合并,然后将合并后的有机相常压蒸馏出二氯甲烷,得到3,3-联二硫酸乙烯酯粗产品。步骤3,将步骤2得到的3,3-联二硫酸乙烯酯粗产品进行蒸馏,以得到精制的3,3-联二硫酸乙烯酯;选择在60℃条件下对3,3-联二硫酸乙烯酯粗产品进行分子蒸馏。实施例4步骤1,以氯化亚砜和赤藓糖醇作为反应原料进行加成反应,以得到中间体3,3-联二亚硫酸乙烯酯;将氯化亚砜加入到第一反应釜中并降温至-10℃,滴加赤藓糖醇,在-10℃条件下充分反应,以得到第一反应液,再将第一反应液进行减压蒸馏,蒸出中间体3,3-联二亚硫酸乙烯酯并通过冷凝回流方式回流到第二反应釜中;氯化亚砜和赤藓糖醇的摩尔比为1.5:1;采用匀速滴加方式向第一反应釜中滴加赤藓糖醇,用时2小时;反应时间为2小时,反应时间为滴加赤藓糖醇的用时和在滴加赤藓糖醇之后继续进行反应的时间;在第一反应釜中进行机械搅拌,以使氯化亚砜和赤藓糖醇充分接触和反应。步骤2,中间体氧化反应得到3,3-联二硫酸乙烯酯;向第二反应釜中加入二氯甲烷和碳酸氢钠并降温至5℃,再向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,在5℃条件下充分反应,得到水相和有机相共存的第二反应液,静置分层后将水相分离出去,得到的有机相即为3,3-联二硫酸乙烯酯粗产品;加入第二反应釜的二氯甲烷的用量,以体积计,为步骤1所得中间体3,3-联二亚硫酸乙烯酯体积的8倍;向第二反应釜中加入碳酸氢钠,以使pH值为8.5;在次氯酸钠和催化剂的水相混合液中,催化剂为二氯化钌,次氯酸钠和催化剂的摩尔比为1:0.0001,次氯酸钠与步骤1所得中间体的摩尔比为6:1,采用匀速滴加方式向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,用时1.5小时;第二反应釜中的反应时间为2.5小时;在第二反应釜中进行机械搅拌,以使各个组份充分接触和反应;水相和有机相共存的第二反应液静置分层进行分液处理,得到水相和有机相,所述的水相用二氯甲烷萃取三次,得到二氯甲烷萃取液,二氯甲烷萃取液与分液处理得到的有机相合并,然后将合并后的有机相常压蒸馏出二氯甲烷,得到3,3-联二硫酸乙烯酯粗产品。步骤3,将步骤2得到的3,3-联二硫酸乙烯酯粗产品进行蒸馏,以得到精制的3,3-联二硫酸乙烯酯;选择在75℃条件下对3,3-联二硫酸乙烯酯粗产品进行分子蒸馏。实施例5步骤1,以氯化亚砜和赤藓糖醇作为反应原料进行加成反应,以得到中间体3,3-联二亚硫酸乙烯酯;将氯化亚砜加入到第一反应釜中并降温至-10℃,滴加赤藓糖醇,在-10℃条件下充分反应,以得到第一反应液,再将第一反应液进行减压蒸馏,蒸出中间体3,3- 联二亚硫酸乙烯酯并通过冷凝回流方式回流到第二反应釜中;氯化亚砜和赤藓糖醇的摩尔比为2.5:1;采用匀速滴加方式向第一反应釜中滴加赤藓糖醇,用时2小时;反应时间为3小时,反应时间为滴加赤藓糖醇的用时和在滴加赤藓糖醇之后继续进行反应的时间;在第一反应釜中进行机械搅拌,以使氯化亚砜和赤藓糖醇充分接触和反应。步骤2,中间体氧化反应得到3,3-联二硫酸乙烯酯;向第二反应釜中加入二氯甲烷和碳酸氢钠并降温至10℃,再向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,在10℃条件下充分反应,得到水相和有机相共存的第二反应液,静置分层后将水相分离出去,得到的有机相即为3,3-联二硫酸乙烯酯粗产品;加入第二反应釜的二氯甲烷的用量,以体积计,为步骤1所得中间体3,3-联二亚硫酸乙烯酯体积的3倍;向第二反应釜中加入碳酸氢钠,以使pH值为7.5;在次氯酸钠和催化剂的水相混合液中,催化剂为二氯化钌,次氯酸钠和催化剂的摩尔比为1:0.00001,次氯酸钠与步骤1所得中间体的摩尔比为2:1,采用匀速滴加方式向第二反应釜中滴加次氯酸钠和催化剂的水相混合液,用时1小时;第二反应釜中的反应时间为1小时;在第二反应釜中进行机械搅拌,以使各个组份充分接触和反应;水相和有机相共存的第二反应液静置分层进行分液处理,得到水相和有机相,所述的水相用二氯甲烷萃取三次,得到二氯甲烷萃取液,二氯甲烷萃取液与分液处理得到的有机相合并,然后将合并后的有机相常压蒸馏出二氯甲烷,得到3,3-联二硫酸乙烯酯粗产品。步骤3,将步骤2得到的3,3-联二硫酸乙烯酯粗产品进行蒸馏,以得到精制的3,3-联二硫酸乙烯酯;选择在65℃条件下对3,3-联二硫酸乙烯酯粗产品进行分子蒸馏。采用现有技术的反应过程进行3,3-联二硫酸乙烯酯的制备,并作为对比例,以氯化亚砜、赤藓糖醇为原料(两者摩尔比为1.2:1),四氯化碳为溶剂,于0℃条件下回流反应30min,然后以高碘酸钠为反应试剂(与赤藓糖醇的摩尔比为2:1),催化剂为三氯化钌(100mg),于0℃条件下反应15min,保温1h,得到3,3-联二硫酸乙烯酯。将上述实施例1—5制备的3,3-联二硫酸乙烯酯进行产品收率、产品纯度、产品酸值和产品水分的测试,产品收率的平均值为92%、产品纯度的平均值为99.8%、产品酸值的平均值为46ppm和产品水分的平均值为16ppm。对依照现有技术制备的3,3-联二硫酸乙烯酯进行产品收率、产品纯度、产品酸值和产品水分的测试,产品收率的平均值为68%、产品纯度的平均值为94%、产品酸值的平均值为600ppm和产品水分的平均值为5000ppm。由此可知,本发明制备的3,3-联二硫酸乙烯酯在纯度、收率、酸值和水分上明 显高于现有技术制备的产品。将本发明制备的3,3-联二硫酸乙烯酯用于电解液中并进行性质测试,电解液的制备方法如下:(1)使用有机溶剂碳酸乙烯酯(EC)、碳酸甲乙酯(EMC)和三氟乙酸乙酯进行混合后,使用4A分子筛、氢化锂除杂,除水;以体积计,碳酸乙烯酯(EC):碳酸甲乙酯(EMC):三氟乙酸乙酯=1:2:0.2;(2)在室温20—25摄氏度下将锂盐溶解在有机溶剂中,锂盐为:LiPF6,锂盐浓度为1mol/L;(3)向有机溶剂中加入添加剂3,3-联二硫酸乙烯酯并分散均匀,即所得锂离子电池电解液,添加剂的质量百分数为1—2wt%。添加剂质量百分数1wt%的电解液和不进行添加的电解液进行测试,将电解液用于NCM(1:1:1)LiNi1/3Co1/3Mn1/3O2/石墨软包电池,测试电池在高温65℃,4.75V1C倍率下的充放电循环性能,结果如附图4所示(65℃循环100周前后的EIS图),添加有添加剂的电池在高温65℃循环50周后,容量保持为90%以上,而未添加本发明添加剂的电池在高温65℃循环50周后,容量仅剩下78%,说明本发明添加剂高温循环性能优越。以上对本发明做了示例性的描述,应该说明的是,在不脱离本发明的核心的情况下,任何简单的变形、修改或者其他本领域技术人员能够不花费创造性劳动的等同替换均落入本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1