通过调节碳代谢途径基因提高S‑腺苷甲硫氨酸产量的方法与流程
本发明属于生物工程领域,更具体地,本发明涉及一种通过调节碳代谢途径基因提高s-腺苷甲硫氨酸产量的方法。
背景技术:
sam普遍存在于动植物和微生物体内,是生物体内一种重要的中间代谢物,参与体内多种生化反应,是由意大利科学家cantoni等发现。sam具有极高的医学价值,对治疗抑郁症、肝脏疾病、关节炎等多种疾病效果显著。因此如何高效、廉价地生产sam是其大规模推广生产的关键。目前主要应用代谢工程的方法获得sam高产菌株以提高sam产量。
迄今为止主要有4条途径可以利用代谢工程的方法来改变酵母的细胞代谢,从而获得sam高产菌。
一是过表达s-腺苷甲硫氨酸合成酶(mat),提高sam合成酶活性促进sam的合成。在生物体内存在两种sam合成酶,甲硫氨酸或sam的存在能够阻止sam1基因的正常表达,但是sam2基因在这种条件下,它可以编码高活性的sam合成酶,并使其表达量显著增加。因此li等(lidy,yuj,tianl,etal.productionofsambyrecombinantpichiapastoris[j].journalofbiotechnology.2002,18:295-299)将sam2基因转入毕赤酵母gs115中,以aox1启动子调控sam2基因的表达,使sam合成酶酶活提高了37倍,sam产量达到了1.58g/l。由于aox1启动子是甲醇诱导型启动子,只有在甲醇存在的情况下才能诱导基因的表达,甘油的存在会对aox1启动子的诱导产生抑制作用,但鉴于甲醇对细胞有毒性,在诱导过程中只能少量添加。因此yu等(yuzl,wuxj,lidy,etal.enhancementoftheproductionofsambyoverexpressionofsamsynthetaseinpichiapastoris[j].actabiochimicaetbiophysicasinica.2003,35:127-132.)尝试用3-磷酸甘油醛脱氢酶(gap)启动子调控sam2基因在毕赤酵母中的表达,使sam产量达到了2.49g/l。hu等(huh,qianjc,chuj,etal.dnashufflingofmethionineadenosyltransferasegeneleadstoimproveds-adenosyl-l-methionineproductioninpichiapastoris[j].journalofbiotechnology.2009,141:97-103.)利用dnashuffling技术将分别来源于链霉菌、酿酒酵母和大肠杆菌的sam合成酶进行了体外进化,筛选出了具有高活性的sam合成酶,将其转入毕赤酵母gs115中获得重组菌ds16,在500l发酵罐上sam的产量达到了6.14g/l。
二是敲除cys4基因(编码胱硫醚β-合成酶),抑制胱硫醚β-合成酶的活性,有效减少sam在胞内向半胱氨酸的转化。he等(hejy,dengjj,zhengyh,etal.asynergisticeffectontheproductionofs-adenosyl-l-methionineinpichiapastorisbyknockinginofs-adenosyl-l-methioninesynthaseandknockingoutofcystathionine-betasynthase[j].journalofbiotechnology.2006,126:519-527.)通过基因敲除的方法将cys4基因敲掉,结果sam积累量提高了两倍多,但是完全阻断sam分解代谢途径,重组菌会成为营养缺陷型,培养基中需外源添加还原型谷胱甘肽(gsh),导致发酵成本大大提高。因此本实验室qin(qinxl,qianjc,yaogf,etal.gappromoterlibraryforfine-tuningofgeneexpressioninpichiapastoris[j].appliedandenvironmentalmicrobiology.2011,77:3600-3608.)利用gap启动子文库弱化cys4基因表达,使其不再是营养缺陷型并能在细胞内大量积累sam,得到一株sam高产菌g12,通过摇瓶培养产量达到了133.2mg/gdcw,在5l罐中重组菌g12的sam产量达到了146.8mg/gdcw。
三是改变甲叉四氢叶酸还原酶(mthfr)对sam的调控,解除其对sam的反馈抑制作用,促进sam的合成。s.cerevisiae中,mthfr有两个同功酶,分别由met12和met13基因编码因此,sanja等(rojes,chansy,kaplanf,etal.metabolicengineeringinyeastdemonstratesthats-adenosylmethioninecontrolsfluxthroughthemethylenetetrahydrofolatereductasereactioninvivo[j].journalofbiologicalchemistry.2002,277:36904.)构建了一个由酵母met13p的n端催化部位以及arabidopsisthalianamthfr(amthfr-1)c端调节部位构成的嵌合mthfr(chimera-1),sam对mthfr的反馈抑制得到解除,基因工程酵母菌scy4的sam产量比野生型菌株提高了140倍。
四是敲除编码△-24-固醇-c-转甲基酶的erg6基因,阻断甾醇合成途径,减少sam的消耗。shobayashi等(shobayashim,mukain,iwashitak,etal.anewmethodforisolationofs-adenosy-l-methionine(sam)-accumulatingyeast[j].appliedmicrobiologyandbiotechnology.2006,69:704-710.)将来源于酿酒酵母k-9和x2180-1a中的erg6基因敲掉,分别获得了来自k-9和x2180-1a的突变株,发酵结果表明来自k-9和x2180-1a突变株的sam产量分别要比出发菌株提高1.7~3倍、4.2~5.5倍。
以上方法仅针对sam合成与降解的局部途径进行改造,具有一定的局限性。本领域技术人员还需利用系统生物学的方法对细胞生理特性进行分析,从全基因组范围寻找影响目标产物合成的改造靶点。
技术实现要素:
本发明的目的在于提供通过调节碳代谢途径基因提高s-腺苷甲硫氨酸产量的方法。
在本发明的第一方面,提供一种生产s-腺苷甲硫氨酸的方法,所述方法包括:
(1)在s-腺苷甲硫氨酸生产菌株的基因组中引入外源的6-磷酸葡萄糖脱氢酶(glucose-6-phosphatedehydrogenase;zwf1)表达盒或6-磷酸葡萄糖酸内酯酶(6-phosphogluconolactonase;sol3)表达盒;
(2)培养步骤(1)制备的菌株,从而生产s-腺苷甲硫氨酸。
在一个优选例中,所述的6-磷酸葡萄糖脱氢酶表达盒包括以下操作性连接的元件:aox1启动子、编码6-磷酸葡萄糖脱氢酶的基因、转录终止序列。
在另一优选例中,所述的6-磷酸葡萄糖酸内酯酶表达盒包括以下操作性连接的元件:aox1启动子、编码6-磷酸葡萄糖酸内酯酶的基因、转录终止序列。
在另一优选例中,所述的在s-腺苷甲硫氨酸生产菌株是(a)插入了sam合成酶基因ds16,并弱化了β-胱硫醚合成酶基因的菌株(例如g12);或(b)在(a)菌株基础上,排除了筛选标记的菌株,例如g12’,其是在g12的基础上用mazf-zeor标记回收单元实现筛选标记的回收,使得新的重组菌g12’不含zeocin抗性,实现了zeor这一标记在毕赤酵母基因操作中的重复利用。
在另一优选例中,所述方法的步骤(2)中,培养的方法包括:30±2℃、220±50rpm条件下进行诱导培养;首次加入甲醇至终浓度1±0.2%后,每隔12±2h补入甲醇至其终浓度为按照体积1±0.2%;每隔24±4h补加l-met,共诱导96±12h。
在本发明的另一方面,提供一种改良的s-腺苷甲硫氨酸生产菌株,所述的菌株的基因组中引入了外源的6-磷酸葡萄糖脱氢酶表达盒或6-磷酸葡萄糖酸内酯酶表达盒。
在一个优选例中,所述的6-磷酸葡萄糖脱氢酶表达盒包括以下操作性连接的元件:aox1启动子、编码6-磷酸葡萄糖脱氢酶的基因,转录终止序列。
在另一优选例中,所述的6-磷酸葡萄糖酸内酯酶表达盒包括以下操作性连接的元件:aox1启动子、编码6-磷酸葡萄糖酸内酯酶的基因、转录终止序列。
在另一优选例中,所述的改良的s-腺苷甲硫氨酸生产菌株是:(a)插入了sam合成酶基因ds16,并弱化了β-胱硫醚合成酶基因的菌株;或(b)在(a)菌株基础上,排除了筛选标记的菌株。
在本发明的另一方面,提供6-磷酸葡萄糖脱氢酶的用途,用于促进s-腺苷甲硫氨酸生产菌株生产s-腺苷甲硫氨酸。
在本发明的另一方面,提供6-磷酸葡萄糖酸内酯酶的用途,用于促进s-腺苷甲硫氨酸生产菌株生产s-腺苷甲硫氨酸。
在本发明的另一方面,提供一种用于生产s-腺苷甲硫氨酸的试剂盒,所述试剂盒中包含所述的改良的s-腺苷甲硫氨酸生产菌株。
本发明的其它方面由于本文的公开内容,对本领域的技术人员而言是显而易见的。
附图说明
图1、质粒paox-zwf1、paox-sol3构建过程示意图。
图2、重组菌g12’/aox-zwf1、g12’/aox-sol3构建过程示意图。对转化子提取基因组dna进行pcr验证并送测序验证。
图3、强化zwf1、sol3基因后,对重组菌生长的影响。
图4、强化zwf1、sol3基因后,对重组菌sam合成的影响。
图5、质粒pcmg(r-a-d)构建过程示意图。
图6、质粒paox-gln构建过程示意图。
图7、质粒paox(ssn)构建过程示意图。
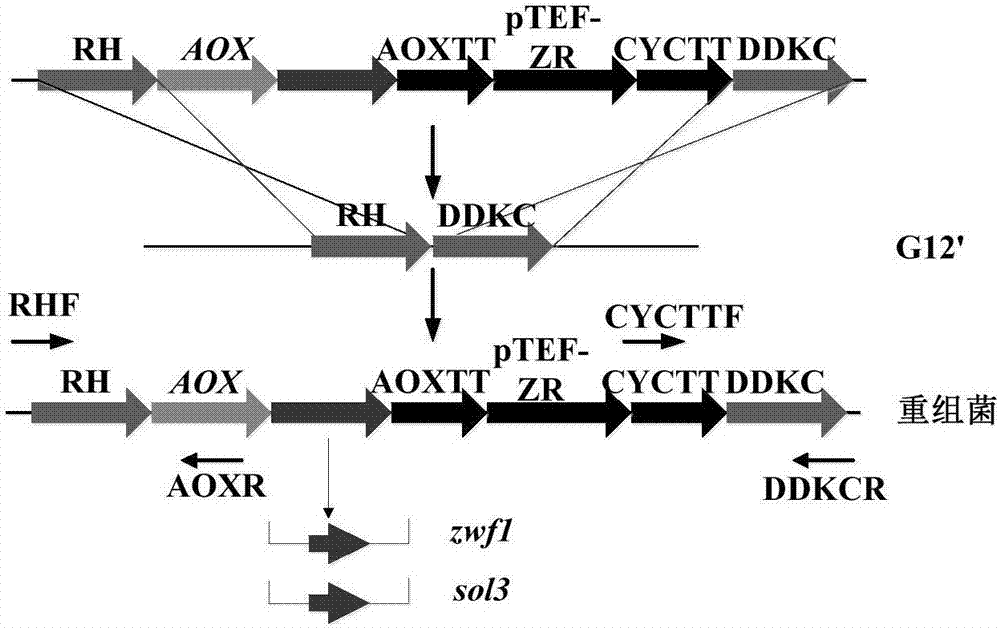
图8、菌株g12’构建的示意图。
具体实施方式
本发明致力于从全基因组范围寻找影响目标产物s-腺苷甲硫氨酸(sam)合成的改造靶点,首次揭示将zwf1或sol3的编码基因转入s-腺苷甲硫氨酸生产菌株,能够显著地提高s-腺苷甲硫氨酸生产菌株的s-腺苷甲硫氨酸产量。
如本文所用,“外源的”或“异源的”是指来自不同来源的两条或多条核酸或蛋白质序列之间的关系。例如,如果启动子与目的基因序列的组合通常不是天然存在的,则启动子对于该目的基因来说是外源的。特定序列对于其所插入的细胞或生物体来说是“外源的”。
如本文所用,所述的“启动子”是指一种核酸序列,其通常存在于目的基因编码序列的上游(5’端),能够引导核酸序列转录为mrna。一般地,启动子或启动子区提供rna聚合酶和正确起始转录所必需的其它因子的识别位点。在本文中,所述的启动子或启动子区包括启动子的活性变异体,该变异体可以是天然发生的等位变异体或非天然发生的变异体。所述变异体包括取代变异体、缺失变异体和插入变异体。
如本文所用,所述的“表达盒”是指包含有表达目的多肽所需的所有必要元件的基因表达系统,通常其包括以下元件:启动子、编码多肽的基因序列,终止子;此外还可选择性包括信号肽编码序列等。这些元件是操作性相连的。
如本文所用,所述的“可操作性连接”是指两个或多个核酸区域或核酸序列的功能性的空间排列。例如:启动子区被置于相对于目的基因核酸序列的特定位置,使得核酸序列的转录受到该启动子区域的引导,从而,启动子区域被“可操作地连接”到该核酸序列上。
ncbi已公开了zwf1多肽序列或zwf1基因序列,因此,本领域人员易于获得和制备所述多肽或基因。作为本发明的优选方式,所述的zwf1具有seqidno:2所示的氨基酸序列,所述的zwf1具有seqidno:1所示的核苷酸序列。
ncbi已公开了sol3多肽序列或sol3基因序列,因此,本领域人员易于获得和制备所述多肽或基因。作为本发明的优选方式,所述的zwf1具有seqidno:4所示的氨基酸序列,所述的zwf1具有seqidno:3所示的核苷酸序列。
在本发明中,术语“zwf1多肽”指具有提高sam产量的功能的seqidno:2序列的多肽;该术语还包括具有提高sam产量的功能的、seqidno:2序列的变异形式。术语“sol3多肽”指具有提高sam产量的功能的seqidno:4序列的多肽;该术语还包括具有提高sam产量的功能的、seqidno:4序列的变异形式。这些变异形式包括(但并不限于):若干个(如1-50个,更佳地1-30个,更佳地1-20个,还更佳如1-10个、1-5个或1-3个)氨基酸的缺失、插入和/或取代,以及在c末端和/或n末端添加或缺失一个或数个(如1-50个,更佳地1-30个,较佳地为1-20个,更佳地为1-10个,如1-5个或1-3个)氨基酸。所述的变异形式均具有提高sam产量的功能。
多肽的变异形式包括:同源序列、保守性变异体、等位变异体、天然突变体、诱导突变体、在高或低的严紧度条件下能与zwf1多肽和/或sol3多肽的dna杂交的dna所编码的多肽。
本发明还涉及zwf1或sol3的多核苷酸变异体,其编码与野生型基因编码的氨基酸序列相同的多肽。此多核苷酸的变异体可以是天然发生的等位变异体或非天然发生的变异体。这些核苷酸变异体包括取代变异体、缺失变异体和插入变异体。如本领域所知的,等位变异体是一个多核苷酸的替换形式,它可能是一个或多个核苷酸的取代、缺失或插入,但不会从实质上改变其编码的多肽的功能。
本发明的zwf1或sol3的核苷酸全长序列或其片段通常可以用pcr扩增法、重组法或人工合成的方法获得。对于pcr扩增法,可根据本发明所公开的有关核苷酸序列,尤其是开放阅读框序列来设计引物,并用市售的cdna库或按本领域技术人员已知的常规方法所制备的cdna库作为模板,扩增而得有关序列。
为了优化s-腺苷甲硫氨酸生产菌株的s-腺苷甲硫氨酸的生产,本发明人作了广泛地研究,从毕赤酵母碳代谢方面进行研究,寻找代谢靶点,并结合基因工程技术构建重组菌,考察队sam合成的影响。sam的合成需要atp提供前体物质,还需要能量和还原力促进菌体生长和维持。在毕赤酵母碳代谢网络中,戊糖磷酸途径(ppp)是nadph等还原力来源的主要途径,本发明人发现,强化这ppp途径有可能提高细胞内的能量和还原力水平,有利于菌体的生长和sam的合成。在此基础上,本发明人找到了适合于进行改良的基因zwf1或sol3,并构建了相应的构建物。
因此,本发明提供了一种构建物,所述构建物包括:zwf1或sol3的表达盒。所述的表达盒具备基因表达所需的所有元件(包括启动子、编码dna以及终止子等),从而可完整地表达出相应的zwf1或sol3多肽。
本领域已知的一些诱导型或组成型启动子均适用于本发明、进行表达盒的构建。作为本发明的优选方式,所述的启动子是甲醇诱导型启动子aox1启动子。当用于增强基因的表达时,一些强启动子是优选的。
通常,所述的构建物位于表达载体上。因此,本发明还包括一种载体,它含有所述的构建物。所述的表达载体通常还含有复制起点和/或标记基因等。本领域的技术人员熟知的方法能用于构建本发明所需的表达载体。这些方法包括体外重组dna技术、dna合成技术、体内重组技术等。所述的dna序列可有效连接到表达载体中的适当启动子上,以指导mrna合成。表达载体还包括翻译起始用的核糖体结合位点和转录终止子。
为了实现在基因组上的基因定向敲除和基因定向重组,所述的表达载体还可设置合适的同源臂;同源臂的设置是本领域技术人员了解的。应理解,只要能够实现对于本发明的s-腺苷甲硫氨酸生产菌株的改造,任何表达载体均可选用的。此外,表达载体优选地包含一个或多个选择性标记基因,以提供用于选择转化的宿主细胞的表型性状。
包含上述的适当多核苷酸序列以及适当启动子或者控制序列的载体,可以用于转化适当的宿主。在本发明的方法中,所述的宿主是s-腺苷甲硫氨酸生产菌株。作为本发明的优选方式,所述的s-腺苷甲硫氨酸生产菌株是酵母菌株,优选地是毕赤酵母菌株,包括各种经改造的毕赤酵母菌株;最优选的,s-腺苷甲硫氨酸生产菌株是g12’菌株,该菌株是以ds16(huh,qianjc,chuj,etal.dnashufflingofmethionineadenosyltransferasegeneleadstoimproveds-adenosyl-l-methionineproductioninpichiapastoris[j].journalofbiotechnology.2009,141:97-103)为出发菌株,g12启动子调控染色体上cbs表达的重组毕赤酵母的菌株。用重组dna转化宿主细胞可用本领域技术人员熟知的常规技术进行。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如j.萨姆布鲁克等编著,《分子克隆指南》,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。
i.材料与方法
1、质粒、菌株和引物
本发明的以下实施例中,应用的质粒如表1所示。
表1、质粒
本发明的以下实施例中,应用的菌株如表2所示。
表2、菌株
本发明的以下实施例中,应用的菌株如表3所示。
表3、引物
表中,下划线表示限制性内切酶切割点。
本发明的以下实施例中,应用的菌株如表4所示。
表4、实验试剂
2、试剂盒与工具酶
所有用于酶切和线性化的限制性内切酶、t4连接酶均购自宝生物工程(大连)有限公司。rtaq酶和pfu酶购自天根生化科技(北京)有限公司。
质粒少量提取、凝胶回收、产物纯化、酵母基因组dna提取试剂盒均购自爱思进生物技术(杭州)有限公司。
3、培养基
(1)大肠杆菌生长培养基(g/l)
lb:酵母提取物10,蛋白胨20,nacl10。
配制含zeocin抗性培养基时zeocin的终浓度为25μg/ml。
配制固体培养基时加2%的琼脂粉。
(2)酵母生长培养基(g/l)
ypd:酵母抽提物10,蛋白胨20,dextrose20。
ypg:酵母抽提物10,蛋白胨20,甘油20。
配制含zeocin抗性培养基时zeocin的终浓度为100μg/ml。
配制固体培养基时加2%的琼脂粉。
(3)酵母摇瓶发酵培养基
bsm:caso40.46,ptm112ml/l,k2so49.1,mgso4·7h2o7.5,(nh4)2so47,0.2mk2hpo4/kh2po4缓冲液(ph=5.5)。
ptm1:h3bo30.02,na2moo4·2h2o0.2,cuso4·5h2o6,znso4·7h2o20,ki0.08,mnso4·h2o3,cocl2·6h2o0.5,feso4·7h2o65,生物素0.2,h2so45ml/l。
4、培养方法
(1)种子培养
从4℃保存的平板上挑取单菌落至3mlypg液体培养基中,30℃、220rpm振荡培养16~18h,取1ml种子液至100mlypg液体培养基中,30℃、220rpm培养20h左右。
(2)摇瓶诱导培养
将种子培养液转移到50ml无菌离心管中,4000rpm离心10min,弃上清。将菌体重悬于含25mlbsm培养基的250ml摇瓶中(使其od600为20左右),30℃、220rpm进行诱导培养。以加入甲醇(1%(v/v))的时间为诱导0h,以后每隔12h补加1%(v/v)的无水甲醇,每隔24h补加630μl40g/l的l-met(已灭菌,终浓度为0.1%),每隔24h测od600的值。培养过程中用5m无菌koh调节ph使其ph保持在5.5-6.0,共诱导96h。
5、检测分析方法
(1)菌体干重的测定
取一定量的菌液稀释一定倍数后利用可见分光光度计在波长600nm处测其吸光值,以无菌的培养基为对照进行比色。od600=od读数×稀释倍数,通过标准曲线计算菌体干重。
菌体干重(drycellweight,dcw)(g/l)=0.2448×od600+1.2349(r2=0.994,od600∈[50,500]。
(2)胞内sam含量测定方法
a.样品处理
取1ml发酵液4℃、12000rpm,离心10min。弃掉上清,用去离子水洗涤一次,重悬于1ml现配的10%的三氯乙酸中,4℃放置抽提2h。然后4℃、12000rpm,离心10min,取上清,用0.22μm的滤膜过滤,4℃保存待检测。浓度大时应稀释适当倍数使hplc中的峰面积在线性范围之内。
b.测定方法
色谱柱为强阳离子型离子交换柱thermo-biobasicscx(型号4.6mm×250mm,5μm)。流动相a为5mm甲酸铵(ph=4.0),流动相b为500mm甲酸铵(ph=4.0)。流速为1ml/min,进样量10μl,柱温30℃,检测波长254nm。sam的保留时间约为10.1min。
(3)胞内sam合成酶酶活测定方法
a.粗酶液的提取
取1ml发酵液,4℃、12000rpm,离心10min。弃掉上清,用去离子水洗涤一次,将所得沉淀重悬于1ml裂解缓冲液中,转入10ml离心管,加入等体积的酸洗玻璃珠(直径为0.5mm),在斡旋振荡器上振荡30s,冰浴30s,重复10次左右后将破碎的菌液转入1.5ml离心管中,4℃、12000rpm,离心10min,取上清即为粗酶液。
b.酶促反应体系
按以上体系建立酶促反应,以不加l-met为空白对照。反应体系在37℃水浴锅中进行。酶反应1h后,加入500μl20%的高氯酸溶液,4℃放置30min以上以停止反应。4℃、12000rpm,离心10min。取上清,用0.22μm的滤膜过滤后检测。
酶活的计算方法:1个酶活单位(u)定义为37℃下1h内转化生成1μmol的sam所对应的sam合成酶的酶活。
实施例1、sol3、zwf1基因强化表达载体构建
paox-ssn构建方法:
依据gs115的基因组序列找到一处可供同源重组的序列,在两个开放阅读框(orf)上各取约700bp作为同源臂,以gs115基因组dna为模板进行pcr获得同源臂,上下游同源臂分别命名为rh和ddkc。用xhoi和spei切除质粒pcmg(乔雪锋,利用无标记基因操作方法优化毕赤酵母合成s-腺苷甲硫氨酸[d].上海:华东理工大学,2013)上的cbs5’片断,将上游同源臂rh连接至原来cbs5’的位置。将padh2-vgb-tt单元(乔雪锋;利用无标记基因操作方法优化毕赤酵母合成s-腺苷甲硫氨酸[d];上海:华东理工大学,2013)和ddkc进行重叠pcr,获取整个大片段。用psti和ecori切除上述已具有rh同源臂的质粒上的pgap-cbsorf片断,将获得的带vgb基因的重叠片断连接至psti与ecori位点之间,得到质粒pcmg(r-a-d)。构建过程如图5。
以质粒pcmg(r-a-d)为出发质粒,通过重叠pcr将aox启动子和gln1基因连接成一个片段,经spei和noti酶切,连接pcmg(r-a-d),得到的质粒命名为paox-gln,如图6。
以paox-gln为出发质粒,经pcr扩增获得带酶切位点的paox,同时用spei和noti酶切片断和质粒,连接得到paox(ssn)。构建过程如图7所示。
以zwf1f和zwf1r为引物,以g12’基因组dna为模板,扩增获得zwf1基因。
以sol3f和sol3r,以g12’基因组dna为模板,扩增获得sol3基因。
以质粒paox-ssn为出发载体,用限制性内切酶sacii和noti对目的基因和质粒paox-ssn双酶切,纯化回收后用t4连接酶连接得到的重组质粒分别命名为paox-zwf1,paox-sol3,构建过程如图1。构建获得的质粒,经酶切和测序验证序列。
实施例2、重组菌构建
将质粒pcmg(乔雪锋.利用无标记基因操作方法优化毕赤酵母合成s-腺苷甲硫氨酸[d];上海:华东理工大学,2013.)经xhoi/ecori双酶切,回收含有mazf-zeor的弱化启动子替换单元,电击转化sam高产菌株ds16。在含有zeocin的平板筛选,得到重组菌g/cmg。然后通过标记回收单元(mazf-zeor)两端正向重复序列之间的同源重组,实现筛选标记的回收,使重组菌g/cmg不再具有zeor,得到新的重组菌g/cg,即g12’(图8)。整个过程还可参见乔雪锋;利用无标记基因操作方法优化毕赤酵母合成s-腺苷甲硫氨酸[d];上海:华东理工大学,2013。
将构建好的重组质粒经xhoi/ecori双酶切线性化,纯化回收目的片段,并将目的片段转入毕赤酵母g12’中,通过rh-ddkc位点,与g12’基因组中的adrh和ddkc发生同源重组整合到g12’基因组上(图2),在含zeocin抗性平板上挑选单克隆。
根据如上步骤,获得重组菌g12’/aox-zwf1和g12’/aox-sol3。
实施例3、重组菌的酶活与产量
将重组菌g12’/aox-zwf1和g12’/aox-sol3进行摇瓶发酵培养,以出发菌株g12’作为对照菌,考察zwf1和sol3基因强化表达对菌体生长以及sam合成的影响。
初始诱导菌浓都控制在同一水平,以补加甲醇为诱导的零点,每隔12h补加甲醇至发酵液中甲醇的终浓度为1%,每隔24h补加1%l-met(w/v),整个过程用5m的koh调节ph在5.5~6.0,共诱导96h。每隔24h取样测od600,得到生长曲线,如图3所示。重组菌的生长趋势与对照菌g12’一致,说明各基因的过表达不影响菌体的正常生长。
诱导培养96h后测定重组菌的sam产量和sam合成酶酶活(图4)。g12’/aox-sol3的sam产量为182.17mg/gdcw,比出发菌g12’(159.11mg/gdcw)提高了14.5%,g12’/aox-zwf1的sam产量为178.87mg/gdcw,相比出发菌提高了12.4%。
考察相应的sam合成酶酶活,g12’/aox-zwf1的酶活最高为442.72u/gdcw,相比出发菌g12’(351.05u/gdcw)提高了26.0%,g12’/aox-sol3的酶活也达到417.62u/gdcw,比出发菌增长了18.7%。
上述结果说明,过表达sol3和zwf1基因能极为显著地提高sam合成酶酶活,促进sam合成。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 利用scmv-cp基因培育抗花叶病甘蔗品种的方法
- 利用SrMV-P<sub>1</sub>基因培育抗花叶病甘蔗品种的方法
- 甜菜素代谢植物酪氨酸酶蛋白及其编码基因与应用的制作方法
- 增加半乳糖分解代谢的基因以及包含所述基因的重组载体和微生物的制作方法
- Mthfr基因snp指导叶酸及相关营养元素摄入的用途、检测方法和试剂盒的制作方法
- 抗代谢类化疗药物疗效相关基因mRNA表达水平检测液相芯片和检测方法
- 一种针对叶酸代谢基因进行基因分型的试剂盒的制作方法
- 用于检测叶酸代谢相关基因snp的引物、荧光探针及应用的制作方法
- 莱茵衣藻油脂代谢基因CrDGAT2-5及其编码蛋白与应用的制作方法
- 参与硝酸盐吸收和代谢的植物基因的制作方法
- 一种维持菌株高效固氮能力的基因的制作方法
- 评价细胞因子对细胞色素p450的代谢能力产生的影响的方法和药剂的筛选方法
- 一种叶酸代谢能力评估的检测位点rs1801131的检测PCR扩增引物和单碱基延伸引物的制作方法
- 一种叶酸代谢能力评估的检测位点rs1801394的检测PCR扩增引物和单碱基延伸引物的制作方法
- 一种用于叶酸代谢能力评估的飞行质谱生物芯片及检测方法和试剂盒的制作方法
- 一种叶酸代谢能力评估的检测位点rs1801133的检测PCR扩增引物和单碱基延伸引物的制作方法
- 血液直接pcr法检测叶酸代谢相关基因多态性的引物、引物组、试剂盒及检测方法
- 一种用于检测汽车防撞杆抗冲击能力的冲击试验的制造方法
- 一种具有适时增粘能力的酸液稠化剂及其制备方法
- 利用scmv-cp基因培育抗花叶病甘蔗品种的方法
- 血液直接pcr法检测叶酸代谢相关基因多态性的引物、引物组、试剂盒及检测方法
- 以二胺为桥基的叶酸修饰荧光染料探针的方法
- 利用scmv-cp基因培育抗花叶病甘蔗品种的方法
- 利用SrMV-P<sub>1</sub>基因培育抗花叶病甘蔗品种的方法
- 甜菜素代谢植物酪氨酸酶蛋白及其编码基因与应用的制作方法
- 增加半乳糖分解代谢的基因以及包含所述基因的重组载体和微生物的制作方法
- Mthfr基因snp指导叶酸及相关营养元素摄入的用途、检测方法和试剂盒的制作方法
- 抗代谢类化疗药物疗效相关基因mRNA表达水平检测液相芯片和检测方法
- 一种针对叶酸代谢基因进行基因分型的试剂盒的制作方法
- 用于检测叶酸代谢相关基因snp的引物、荧光探针及应用的制作方法