腐食酪螨新发现过敏原基因克隆表达纯化及其应用的制作方法
本发明属于分子生物学领域,具体涉及腐食酪螨新过敏原Tyrp28、Tyrp35、Tyrp36的原核表达纯化及其应用。
背景技术:
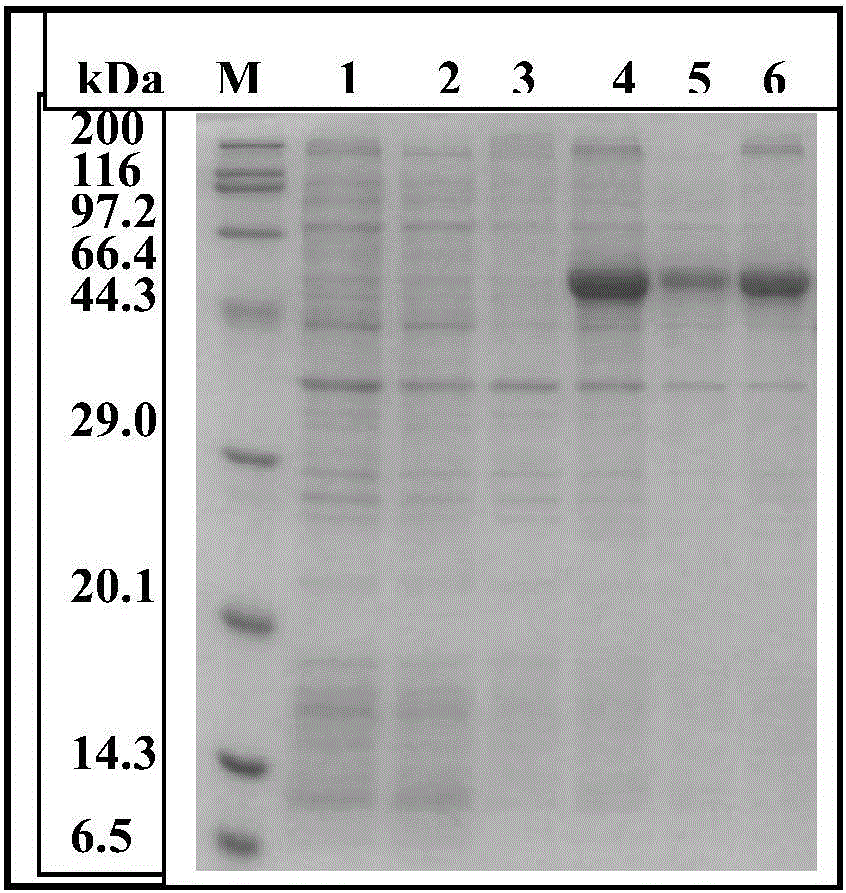
:螨虫问题越来越严重,引起了人们的高度关注!专家提示:世界上每五个人中就至少有一人会出现过敏。而尘螨是导致过敏的罪魁祸首。别小看了这些身长只有0.2—0.4mm的小虫子,它们能够引发的疾病可绝对不少呢。甚至会引发螨虫过敏性哮喘,直接威胁生命。根据研究调查显示,过敏性鼻炎患者和非过敏性鼻炎患者得哮喘的风险分别是常人的3.5倍和2.7倍。过敏是引起儿童哮喘的一个重要因素,我国的儿童过敏性哮喘患者目前有800多万,当中有80%都是由尘螨引起的。腐食酪螨是一种重要的过敏原来源螨种。对100例英国支气管哮喘患者进行皮肤挑刺试验,有34%例对腐食酪螨呈阳性反应;对72例伦敦支气管哮喘/鼻炎患者进行皮肤挑刺试验,51%例对腐食酪螨呈阳性反应;50%的东南亚支气管哮喘患者对腐食酪螨呈阳性反应(ArlianLG,GeisDP,Vyszenski-MoherDL,BernsteinIL,GallagherJS.CrossantigenicandallergeicpropertiesofthehousedustmiteDermatophagoidesfarinaeandthestoragemiteTyrophagusputrescentiae.JAllergyClinImmunol.1984,74:172-179.)。但是临床诊断依靠进口的腐食酪螨粗提浸液混合物诊断过敏性疾病患者,其中含有过敏原、非过敏原、甚至有毒蛋白,而患者病因可能只与粗提浸液中的某种或某些变应原有关,这就意味着该粗提浸液特异性差。采用基因工程技术获得过敏原单一组分,其成分单一,纯度高、特异性好,可以提高诊断的精准性。因此,如何提供一种腐食酪螨过敏原基因克隆、表达、纯化的方法,提高产物的纯度,提高阳性率是本领域技术人员亟待解决的技术问题。技术实现要素:针对现有技术中的不足,本发明进行了深入研究,提供了一种一种腐食酪螨新发现过敏原基因克隆表达纯化方法。本发明一方面涉及一种腐食酪螨新发现过敏原基因克隆表达纯化方法,其包括如下步骤:1目的基因获取1.1引物设计和合成以腐食酪螨TotalRNA为模板,RT-PCR扩增KX060612序列CDS区1461bp克隆至pET28a(+)载体;1.2反转录和PCR扩增,并使用TaKaRaMiniBESTAgaroseGelDNAExtractionKitVer.4.0(CodeNo.9762)切胶回收目的产物,命名为CTG0124-PCR;2克隆引物设计和合成:名称序列(5′-3′)长度(mers)CTG0124P1CACTGATGCTCGCCTGGAAG20使用HDCloningKit(ClontechCodeNo.639633),将CTG0124-PCR和VectorDNA2连接;热转化至E.coliCompetentCellsJM109(CodeNo.9052)中,涂布平板,37℃过夜培养。使用T7/T7terminator引物挑选阳性菌落植菌,提取阳性克隆质粒CTG0124-1;将质粒CTG0124-1转入BL21(DE3)T1R感受态细胞中,对阳性克隆进行诱导表达和培养;挑取单菌落至2mLLB/Amp(100μg/mL)培养基中,37℃O/N培养,命名为EXP0212-2,添加IPTG进行诱导,37℃培养,集菌前吸光度值为2.65;集菌后进行超声波破碎,对菌体破碎液进行离心分离;使用EXP0212-2甘油菌保进行500mL培养,以从菌体中进行目的蛋白纯化:使用GEHiTrapTALONcrude,5mLTALONSuperflow(CodeNo.28-9537-66)柱层析和;使用10倍柱体积的灭菌MilliQH2O50mL和10倍柱体积的BufferA50mL平衡树脂,流速1mL/min;上样:BufferA平衡化后上样,流速为0.5mL/min;蛋白吸附后,使用10倍体积的BufferA50mL清洗树脂,流速1mL/min,W1:10mL,W2:20mL,W3:20mL;蛋白溶出Elution:BufferA50mL、BufferB50mL梯度溶出,流速1mL/min,Fraction1.1mL/tube×90tubes;收集目的蛋白FractionNo.46~90,使用Nanodrop-100进行定量分析,获得纯化重组蛋白,蛋白纯度为99%。本发明还涉及上述腐食酪螨新发现过敏原基因克隆表达纯化方法所得到的重组蛋白。本发明还涉及上述重组蛋白在制备诊断试剂中的应用,优选的,所述的诊断试剂用于腐食酪螨过敏源临床诊断。本申请公开了一种腐食酪螨新发现过敏原基因克隆表达纯化方法,本发明的方法特定的引物和特定的纯化方法所得到的纯化产物纯度高,阳性率高,能够作为临床诊断试剂使用,具有广泛的应用前景。附图说明图1腐食酪螨转录组序列CL2629.Contig1_RNA_1A全长基因PCR后琼脂糖凝胶电泳图。LaneM,DL2,000DNAMarker;Lane1,PCR产物。图2EXP0212-2表达SDS-PAGE图像。图3EXP0212-2的PVDF图像。LaneM1,PrecisionPlusProtein;Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0212-2全细胞;Lane5,EXP0212-2上清;Lane6,EXP0212-2沉淀。图4500mLEXP0230表达的SDS-PAGE图像。图5:LaneM,proteinMWmarker(Broad);Lane1,EXP0230S51μL上样;Lane2,EXP0230S52μL上样;Lane3,EXP0230S54μL上样;Lane4,BSA200ng;Lane5,BSA400ng;Lane6,BSA600ng;Lane7,BSA800ng;Lane8,BSA1000ng。图6:Westernblotting检测结果。图7-腐食酪螨转录组序列Unigene7127_RNA_1A全长基因PCR后琼脂糖凝胶电泳图。LaneM,DL2,000DNAMarker;Lane1,PCR产物。图8-EXP0207-2表达SDS-PAGE图像(15%polyacrylamidegel)LaneM,ProteinMWmarker(Broad);Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0207-2全细胞;Lane5,EXP0207-2上清;Lane6,EXP0207-2沉淀。图9-EXP0207-2的PVDF图像LaneM1,PrecisionPlusProteinStandards;LaneM2,perfectproteinmarker;Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0207-2全细胞;Lane5,EXP0207-2上清;Lane6,EXP0207-2沉淀。图10-500mLEXP0228表达的SDS-PAGE图像LaneM,ProteinMWmarker(Broad);Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0228全细胞;Lane5,EXP0228上清;Lane6,EXP0228沉淀;Lane7,BSA200ng;Lane8,BSA500ng;Lane9,BSA1000ng;Lane10,BSA1500ng;Lane11,BSA2000ng。图11:LaneM,proteinMWmarker(Broad);Lane1,EXP0228S51L上样;Lane2,EXP0228S52L上样;Lane3,EXP0228S54L上样;Lane4,BSA200ng;Lane5,BSA400ng;Lane6,BSA600ng;Lane7,BSA800ng;Lane8,BSA1000ng。图12Westernblotting检测结果。图13腐食酪螨TotalRNA琼脂糖凝胶电泳图。LaneM,Marker;Lane1,腐食酪螨TotalRNA。图14腐食酪螨转录组序列CL2396.Contig1_RNA_1A全长基因PCR后琼脂糖凝胶电泳图。LaneM,DL2,000DNAMarker;Lane1,PCR产物。图15-EXP0212-1表达的SDS-PAGE图像:LaneM,proteinMWmarker(Broad);Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane2,pET28a(+)沉淀;Lane4,EXP0212-1全细胞;Lane5,EXP0212-1上清;Lane6,EXP0212-1沉淀。图16EXP0212-1PVDF膜图像:LaneM1,PrecisonPlusProteinStandards;Lane1,pET-28a(+)全细胞;Lane2,pET-28a(+)上清;Lane3,pET-28a(+)沉淀;Lane4,EXP0212-1全细胞;Lane5,EXP0212-1上清;Lane6,EXP0212-1沉淀;LaneM2,perfectproteinmarker。图17EXP0231500ml表达SDS-PAGE图像:LaneM,ProteinMWmarker(Broad);Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0231全细胞;Lane5,EXP0231上清;Lane6,EXP0231沉淀;Lane7,BSA200ng;Lane8,BSA500ng;Lane9,BSA1000ng;Lane10,BSA1500ng;Lane11,BSA2000ng。图18透析后样品EXP0231-S5的SDS-PAGE电泳图:LaneM,ProteinMWmarker(Broad);Lane1,1L上样EXP0231S5;Lane2,2L上样EXP0231S5;Lane3,4L上样EXP0231S5;Lane4,BSA200ng;Lane5,BSA400ng;Lane6,BSA600ng;Lane7,BSA800ng;Lane8,BSA1000ng。图19EXP0231-S5重组蛋白Westernblotting检测结果:1-17为阳性血清,阳性率为47%(8/17)。具体实施方式下面结合实施例对本发明做进一步的说明,以下所述,仅是对本发明的较佳实施例而已,并非对本发明做其他形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更为同等变化的等效实施例。凡是未脱离本发明方案内容,依据本发明的技术实质对以下实施例所做的任何简单修改或等同变化,均在本发明的保护范围内。实施例1:腐食酪螨过敏原Tryp35基因克隆表达纯化及其应用全长基因扩增、克隆及原核表达质粒构建1目的基因获取1.1引物设计和合成以腐食酪螨TotalRNA为模板,RT-PCR扩增KX060612序列CDS区1461bp克隆至pET28a(+)载体。1.2反转录使用3′-FullRACECoreSetwithPrimeScriptTMRTase(CodeNo.6106)合成cDNA,同时设立M-MLV(-)对照。反应体系:1μgTotalRNA、1μLRandom6(20μM)、1μLdNTPMixture(10mMeach)、加dH2ORNaseFree至5μL。反应条件:70℃、10min,然后立即冰上放置2min。然后加入下列组分:2μL5×M-MLVBuffer、0.25μLRNaseInhibitor(40U/μL)、0.25μLReverseTranscriptaseM-MLV(RNaseH-)(200U/μL),总反应体积10μL。反应条件:30℃,10min→42℃,60min→70℃,15min。1.3PCR扩增使用TaKaRaTksGflexDNAPolymerase(CodeNo.R060A)进行PCR扩增。反应体系:2μL反转录反应液、25μL2×GflexPCRBuffer(Mg2+,dNTPplus)、1μLTksGflexDNAPolymerase(1.25units/μL)、1μLCTG0124FPrimer(10μM)、1μLCTG0124RPrimer(10μM)、20μLdH2O,总反应体积50μL。反应条件:94℃预变性1min,然后置98℃10sec、55℃15sec、68℃2min反应30个循环。取5μL进行3%琼脂糖凝胶电泳,结果见图1。1.4PCR产物纯化使用TaKaRaMiniBESTAgaroseGelDNAExtractionKitVer.4.0(CodeNo.9762)切胶回收目的产物,命名为CTG0124-PCR。2克隆测序引物设计和合成:名称序列(5′-3′)长度(mers)CTG0124P1CACTGATGCTCGCCTGGAAG20使用HDCloningKit(ClontechCodeNo.639633),将CTG0124-PCR和VectorDNA2(CTG0009项目制备)连接;热转化至E.coliCompetentCellsJM109(CodeNo.9052)中,涂布平板,37℃过夜培养。使用T7/T7terminator引物挑选阳性菌落植菌,提取阳性克隆质粒CTG0124-1、2。使用T7、T7terminator、CTG0124P1引物对上述质粒进行测序。获得质粒CTG0124-1。2.目的蛋白表达及Westernblotting鉴定将质粒CTG0124-1转入BL21(DE3)T1R感受态细胞中,对阳性克隆进行诱导表达。2.1转化取1μL质粒CTG0124-1转化CompetentcellBL21(DE3)T1R中,使用LB/抗生素Amp(100μg/mL)平板,35μL转化液涂布,37℃O/N培养,ControlpET28a(+)进行同样操作。2.2培养及诱导2.2.1种培养分别挑取单菌落至2mLLB/Amp(100μg/mL)培养基中,37℃O/N培养,命名为EXP0212-2。2.2.2主培养诱导分别在Glasstube中添加5mLLB/Amp(100μg/mL)培养基中,添加种培养菌液100μL。37℃培养至OD600nm值约为0.6,添加100mMIPTG50μL(final1mMIPTG)进行诱导,37℃培养4h。诱导前吸光度值为0.60,集菌前吸光度值为2.65。2.2.3蛋白质抽提集菌后2.0OD相当的菌体加入320μLPBS悬浊后进行超声波破碎,对菌体破碎液进行离心分离(12000×rpm、5min)。2.2.4抽提液电泳取抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min,使用12.5%polyacrylamidegel,电泳结束后,CBB-R250染色,使用脱色液脱色。SDS-PAGE/CBB染色结果见图2。LaneM,ProteinMWmarker(Broad);Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0212-2全细胞;Lane5,EXP0212-2上清;Lane6,EXP0212-2沉淀。2.3Westernblotting取抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,使用彩色Marker及组氨酸标签Marker,进行SDS-PAGE电泳,电泳条件:25mA/枚,80min,使用12.5%polyacrylamidegel。电泳结束后,将PVDF膜、滤纸分别剪切成与凝胶相同大小,使用转膜缓冲液处理后,按滤纸、PVDF膜、凝胶、滤纸的顺序依次放在转膜仪电极板之间,开始转膜,设置转膜仪参数如下:电流50mA、转印时间80min。将PVDF膜置于含1.5%BSA的10mLBlockingbuffer中,4℃平放过夜封闭。使用稀释后的Pena-HisAntibody溶液5mL,进行一次抗体反应1h。TBST缓冲液(20mL)洗涤2次,洗涤TBS缓冲液冲洗3次。使用稀释后的HRP-RabbitAnti-MouseIgG抗体溶液5mL,进行二次抗体反应1h。TBST缓冲液(20mL)洗涤2次;洗涤TBS缓冲液冲洗3次。1mLTrueBluePeroxidaseSubstrate显色1min。显色后PVDF膜见图3。由图可见质粒EXP0212-2(CTG0124-1)为部分可溶性表达。3.目的蛋白纯化及鉴定使用EXP0212-2甘油菌保进行500mL培养,以从菌体中进行目的蛋白纯化。3.1500mL培养3.1.1种培养取EXP0212-2样品甘油菌保质粒50μL植入10mLLB/Kana(50μg/mL)培养基中,37℃O/N培养。3.1.2主培养及诱导将10mL种培养菌液植入500mLLB/2LFlask中,37℃、200rpm培养至OD600约0.6时,添加100mMIPTG500μL(final0.1mMIPTG)进行诱导,16℃继续培养24h。培养后离心,收集菌体。使用PBS清洗菌体后离心,称量菌体湿重。诱导前吸光度为了0.60、集菌前吸光度为3.75,菌体湿重为1.72g。3.1.3SDS-PAGE取2.0OD相当的菌体加入320μLPBS悬浊后进行超声波破碎,对菌体破碎液进行离心分离(12000×rpm、5min)。取抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min。使用12.5%polyacrylamidegel,电泳结束后,CBB-R250染色,使用脱色液脱色。SDS-PAGE/CBB染色结果见图4。SDS-PAGE结果表达500mL培养菌体有表达,主要为可溶性表达。LaneM,ProteinMWmarker(Broad);Lane1,pET28a(+)全细胞;Lane2,pET28a(+)上清;Lane3,pET28a(+)沉淀;Lane4,EXP0230全细胞;Lane5,EXP0230上清;Lane6,EXP0230沉淀;Lane7,BSA200ng;Lane8,BSA500ng;Lane9,BSA1000ng;Lane10,BSA1500ng;Lane11,BSA2000ng。3.2重组蛋白纯化3.2.1缓冲液配制Sonicationbuffer/Washbuffer/BufferA,pH8.0(4℃),由50mMSodiumphosphate(pH8.0)、300mMNaCl、5mMImidazole组成;BufferB,pH8.0(4℃),由50mMSodiumphosphate(pH8.0)、300mMNaCl、300mMImidazole组成。透析Buffer为TaKaRaPBS(CodeNo.T900)。3.2.2菌体破碎培养后菌体湿重为1.72g,加入Sonicationbuffer(菌体湿重的10倍)20mL;4℃充分悬浊,Sonication180w,10min,收集破碎液20mL(S1);将S1置12000rpm、4℃离心30min、得到上清20mL(S2),沉淀0.2g;取S2使用0.45μm、1000mLVacuumfilter/Storagebottlesystem进行过滤,得到样品20mL(S3)。3.2.3柱层析纯化使用GEHiTrapTALONcrude,5mLTALONSuperflow(CodeNo.28-9537-66)柱层析和;使用10倍柱体积的灭菌MilliQH2O50mL和10倍柱体积的BufferA50mL平衡树脂,流速1mL/min。上样:BufferA平衡化后上样,流速为0.5mL/min。蛋白吸附后,使用10倍体积的BufferA50mL清洗树脂,流速1mL/min,W1:10mL,W2:20mL,W3:20mL。蛋白溶出Elution:BufferA50mL、BufferB50mL梯度溶出,流速1mL/min,Fraction1.1mL/tube×90tubes。3.2.4各阶段SDS-PAGE电泳及AKTA出峰图各纯化各阶段进行SDS-PAGE电泳,取全细胞(W)、上清(S)、不溶性沉淀(P)各0.05OD600电泳,取S1、S2、S3、Pa原液各1μL电泳,取W1、W2、W3原液各2.5μL电泳,取Elution原液各5μL电泳;取BSA标准品500ng上样。Proteinmarker:BroadWeightMarker(TaKaRaCode:3452Q),5μL/well。电泳胶为12.5%SDS-PAGE/考马斯亮蓝染色液染。3.2.5收集收集目的蛋白FractionNo.46~90共计约48mL[S4]。取小量进行SDS-PAGE电泳并以BSA标准品浓度为参考。使用12.5%SDS-PAGE/考马斯亮蓝染色液染色。3.2.6透析使用PBS进行Buffer透析,反复进行3次,透析后样品为[S5]。透析Buffer为PBS,置换率为1:1000,透析膜:SnakeSkinDialysisTubing3500MWCO(CatNo.68035,LotNo.MH160055),使用12.5%SDS-PAGE/考马斯亮蓝染色液染色,取透析后样品进行SDS-PAGE电泳,结果如图5所示。使用Nanodrop-100进行定量分析,获得纯化重组蛋白50mL,浓度为1.30mg/mL,共65mg,蛋白纯度为99%。4.Westernblotting检测取重组蛋白8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚,70min。使用12.5%polyacrylamidegel,电泳结束后,CBB-250染色,使用脱色液脱色。将PVDF膜、滤纸分别剪切成与凝胶相同大小,使用转膜缓冲液处理后,按滤纸、PVDF膜、凝胶、滤纸的顺序依次放在转膜仪电极板之间,2张膜同时开始转膜,设置转膜仪参数如下:电流52mA、转印时间80min。将PVDF膜置于含1.5%BSA的12mLBlockingbuffer中,37℃平放1h封闭。使用1:10稀释后的血清溶液10mL,进行一次抗体反应1h。TBST缓冲液(25mL)洗涤1次;洗涤TBS缓冲液冲洗2次。使用1:2500稀释后的GoatAnti-humanIgE(HRP)抗体溶液9mL进行二次抗体反应1h。TBST缓冲液(25mL)洗涤1次;洗涤TBS缓冲液冲洗2次。1mLTrueBluePeroxidasesubstrate显色1min。显色后PVDF膜如图6所示,结果显示本发明的重组蛋白具有较高的阳性检出率。实施例2:腐食酪螨过敏原Tryp36基因克隆表达纯化及WB验证1.全长基因扩增、克隆及原核表达质粒构建以腐食酪螨TotalRNA为模板,RT-PCR扩增GenBankNo.KX060615序列CDS区396bp克隆至pET28a(+)载体。1.1目的基因获取1.1.1引物设计和合成1.1.2反转录使用3′-FullRACECoreSetwithPrimeScriptTMRTase(CodeNo.6106)合成cDNA,同时设立M-MLV(-)对照。反应体系:1μgTotalRNA、1μLRandom6(20μM)、1μLdNTPMixture(10mMeach)、加dH2ORNaseFree至7.5μL。反应条件:70℃、10min,然后立即冰上放置2min。然后加入下列组分:2μL5×M-MLVBuffer、0.25μLRNaseInhibitor(40U/μL)、0.25μLReverseTranscriptaseM-MLV(RNaseH-)(200U/μL),总反应体积10μL。反应条件:30℃,10min→42℃,60min→70℃,15min。1.1.3PCR扩增使用TaKaRaTksGflexDNAPolymerase(CodeNo.R060A)进行PCR扩增。反应体系:2μL反转录反应液、25μL2×GflexPCRBuffer(Mg2+,dNTPplus)、1μLTksGflexDNAPolymerase(1.25units/μL)、1μLCTG0071FPrimer(10μM)、1μLCTG0071RPrimer(10μM)、20μLdH2O,总反应体积50μL。反应条件:94℃预变性1min,然后置98℃10sec、55℃15sec、68℃1min反应30个循环。取5μL进行3%琼脂糖凝胶电泳,结果见图7。1.1.4PCR产物纯化使用TaKaRaMiniBESTAgaroseGelDNAExtractionKitVer.4.0(CodeNo.9762)切胶回收目的产物,命名为CTG0071-PCR。1.1.5测序分析使用引物CTG0057F1/R0对CTG0071-PCR产物进行测序。1.2克隆测序使用HDCloningKit(ClontechCodeNo.639633),将CTG0071-PCR和VectorDNA1(CTG0006项目制备)连接;热转化至E.coliCompetentCellsJM109(CodeNo.9052)中,涂布平板,37℃过夜培养。使用T7/T7terminator引物挑选阳性菌落植菌,提取阳性克隆质粒CTG0071-1、2。使用T7对上述质粒进行测序。获得质粒CTG0071-1符合要求。2.目的蛋白表达及Westernblotting鉴定将质粒CTG0071-1转入BL21(DE3)T1R感受态细胞中,对阳性克隆进行诱导表达。2.1转化取1μL质粒CTG0071-1转化CompetentcellBL21(DE3)T1R中,使用LB/抗生素Amp(100μg/mL)平板,35μL转化液涂布,37℃O/N培养,ControlpET28a(+)进行同样操作。2.2培养及诱导2.2.1种培养分别挑取单菌落至2mLLB/Amp(100μg/mL)培养基中,37℃O/N培养,命名为EXP0207-2。2.2.2主培养诱导分别在Glasstube中添加5mLLB/Amp(100μg/mL)培养基中,添加种培养菌液100μL。37℃培养至OD600nm值约为0.6,添加100mMIPTG50μL(final1mMIPTG)进行诱导,37℃培养4h。EXP0207-2(CTG0070-73)诱导前吸光度为0.60、集菌前吸光度为1.60,对照pET28a(+)诱导前吸光度值为0.60、集菌前吸光度值为1.35。2.2.3蛋白质抽提集菌后2.0OD相当的菌体加入320μLPBS悬浊后进行超声波破碎,对菌体破碎液进行离心分离(12000×rpm、5min)。2.2.4抽提液电泳取抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min,使用15%polyacrylamidegel,电泳结束后,CBB-R250染色,使用脱色液脱色。SDS-PAGE/CBB染色结果见图8。2.3Westernblotting取抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,使用彩色Marker及组氨酸标签Marker,进行SDS-PAGE电泳,电泳条件:25mA/枚,80min,使用15%polyacrylamidegel。电泳结束后,将PVDF膜、滤纸分别剪切成与凝胶相同大小,使用转膜缓冲液处理后,按滤纸、PVDF膜、凝胶、滤纸的顺序依次放在转膜仪电极板之间,开始转膜,设置转膜仪参数如下:电流39mA、转印时间80min。将PVDF膜置于含1.5%BSA的10mLBlockingbuffer中,4℃平放过夜封闭。使用稀释后的Pena-HisAntibody溶液5mL,进行一次抗体反应1h。TBST缓冲液(20mL)洗涤2次,洗涤TBS缓冲液冲洗3次。使用稀释后的HRP-RabbitAnti-MouseIgG抗体溶液5mL,进行二次抗体反应1h。TBST缓冲液(20mL)洗涤2次;洗涤TBS缓冲液冲洗3次。1mLTrueBluePeroxidaseSubstrate显色1min。显色后PVDF膜见图9。由图可见质粒EXP0207-2(CTG0071-1)主要为可溶性表达。3.目的蛋白纯化及鉴定使用EXP0207-2甘油菌保进行500mL培养,以从菌体中进行目的蛋白纯化。公司命名为EXP0228。3.1500mL培养3.1.1种培养取EXP0207-2样品甘油菌保质粒50μL植入10mLLB/Kana(50μg/mL)培养基中,37℃O/N培养。3.1.2主培养及诱导将10mL种培养菌液植入500mLLB/2LFlask中,37℃、200rpm培养至OD600约0.6时,添加100mMIPTG500μL(final0.1mMIPTG)进行诱导,继续培养4h。培养后离心,收集菌体。使用PBS清洗菌体后离心,称量菌体湿重。诱导前吸光度为了0.60、集菌前吸光度为1.95,菌体湿重为1.15g。3.1.3SDS-PAGE取2.0OD相当的菌体加入320μLPBS悬浊后进行超声波破碎,对菌体破碎液进行离心分离(12000×rpm、5min)。取抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min。使用15%polyacrylamidegel,电泳结束后,CBB-R250染色,使用脱色液脱色。SDS-PAGE/CBB染色结果见图10。SDS-PAGE结果表达500mL培养菌体有表达,基本为可溶性表达。4.2重组蛋白纯化4.2.1缓冲液配制Sonicationbuffer/Washbuffer/BufferA,pH8.0(4℃),由50mMSodiumphosphate(pH8.0)、300mMNaCl、5mMImidazole组成;BufferB,pH8.0(4℃),由50mMSodiumphosphate(pH8.0)、300mMNaCl、300mMImidazole组成。透析Buffer为TaKaRaPBS(CodeNo.T900)。4.2.2菌体破碎培养后菌体湿重为1.15g,加入Sonicationbuffer(菌体湿重的10倍)10mL;4℃充分悬浊,Sonication180w,10min,收集破碎液10mL(S1);将S1置12000rpm、4℃离心30min、得到上清15mL(S2),沉淀0.31g;取S2使用0.45μm、1000mLVacuumfilter/Storagebottlesystem进行过滤,得到样品10mL(S3)。4.2.3柱层析纯化使用GEHiTrapTALONcrude,5mLTALONSuperflow(CodeNo.28-9537-66)柱层析;使用10倍柱体积的灭菌MilliQH2O50mL和10倍柱体积的BufferA50mL平衡树脂,流速1mL/min。上样:BufferA平衡化后上样,流速为0.5mL/min。蛋白吸附后,使用10倍体积的BufferA50mL清洗树脂,流速1mL/min,W1:10mL,W2:20mL,W3:20mL。蛋白溶出Elution:BufferA50mL、BufferB50mL梯度溶出,流速1mL/min,Fraction1.1mL/tube×90tubes。4.2.4各阶段SDS-PAGE电泳及AKTA出峰图各纯化各阶段进行SDS-PAGE电泳,取全细胞(W)、上清(S)、不溶性沉淀(P)各0.05OD600电泳,取S1、S2、S3、Pa原液各1μL电泳,取W1、W2、W3原液各2.5μL电泳,取Elution原液各5μL电泳;取BSA标准品500ng上样。Proteinmarker:BroadWeightMarker(TaKaRaCode:3452Q),5μL/well。电泳胶为15%SDS-PAGE/考马斯亮蓝染色液染。4.2.5收集收集目的蛋白FractionNo.40~65共计约28mL[S4]。取小量进行SDS-PAGE电泳并以BSA标准品浓度为参考。使用15%SDS-PAGE/考马斯亮蓝染色液染色。4.2.6透析使用PBS进行Buffer透析,反复进行3次,浓缩后样品为[S5]。透析Buffer:PBS。置换率:1:1000。透析膜:SnakeSkinDialysisTubing10000MWCO(CatNo.68035,LotNo.MH1600)。使用15%SDS-PAGE/考马斯亮蓝染色液染色,取透析浓缩后样品进行SDS-PAGE电泳,如图11所示。使用Nanodrop-1000进行定量分析:共获得EXP0228-S5,体积30mL,蛋白浓度为:0.94mg/mL,蛋白总量为28.2mg,蛋白纯度为99%。4.Westernblotting检测取重组蛋白8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚,70min。使用12.5%polyacrylamidegel,电泳结束后,CBB-250染色,使用脱色液脱色。将PVDF膜、滤纸分别剪切成与凝胶相同大小,使用转膜缓冲液处理后,按滤纸、PVDF膜、凝胶、滤纸的顺序依次放在转膜仪电极板之间,2张膜同时开始转膜,设置转膜仪参数如下:电流52mA、转印时间80min。将PVDF膜置于含1.5%BSA的12mLBlockingbuffer中,37℃平放1h封闭。使用1:10稀释后的血清溶液10mL,进行一次抗体反应1h。TBST缓冲液(25mL)洗涤1次;洗涤TBS缓冲液冲洗2次。使用1:2500稀释后的GoatAnti-humanIgE(HRP)抗体溶液9mL进行二次抗体反应1h。TBST缓冲液(25mL)洗涤1次;洗涤TBS缓冲液冲洗2次。1mLTrueBluePeroxidasesubstrate显色1min。显色后PVDF膜如图12所示。实施例3:腐食酪螨过敏原Tryp28基因克隆表达纯化及WB验证1.TotalRNA提取提取提供的置于Trizol中的腐食酪螨TotalRNA,然后使用RecombinantDNaseⅠ(RNaseFree)(CodeNo.2270A)进行DNaseⅠ处理,最终溶于DEPC处理水。取1μL进行1%琼脂糖凝胶电泳,结果如图13所示。2.全长基因扩增、克隆及原核表达载体构建以腐食酪螨TotalRNA为模板,RT-PCR扩增GenBankNo.KX060611序列CDS区1980bp克隆至pET28a(+)载体。2.1目的基因获得2.1.1引物设计和合成2.1.2反转录使用3′-FullRACECoreSetwithPrimeScriptTMRTase(CodeNo.6106)合成cDNA,同时设立M-MLV(-)对照。反应体系:1μgTotalRNA、1μLRandom6(20μM)、1μLdNTPMixture(10mMeach),加RNaseFreedH2O至7.5μL。反应条件:70℃10min,立即冰上放置2分钟。然后加入下列组分:2μL5×M-MLVBuffer、0.25μLRNaseInhibitor(40U/μL)、0.25μLReverseTranscriptaseM-MLV(RNaseH-)(200U/μL),总体积10μL。反应条件:30℃反应10min、42℃反应60min、70℃反应15min。2.1.3PCR扩增使用TaKaRaTksGflexDNAPolymerase(CodeNo.R060A),进行PCR扩增。反应体系:2μL上述反转录反应液、25μL2×GflexPCRBuffer(Mg2+,dNTPplus)、1μLTksGflexDNAPolymerase(1.25units/μL)、1μLCTG0123FPrimer(10μM)、1μLCTG0123RPrimer(10μM)、20μLdH2O,总反应体积50μL。反应条件:94℃预变性1min,98℃变性10sec、55℃退火15sec、68℃延伸2min,30个循环。取5μL进行3%琼脂糖凝胶电泳,结果见图14。使用TaKaRaMiniBESTAgaroseGelDNAExtractionKitVer.4.0(CodeNo.9762)切胶回收目的产物,命名为CTG0123-PCR。2.2基因全长克隆测序2.2.1引物设计和合成名称序列(5′-3′)长度(mers)CTG0123P1TCAACGACAGCCAGCGACAG20CTG0123P4TCCTTCACCATGCGTTCAAT202.2.2PCR产物克隆及测序使用HDCloningKit(ClontechCodeNo.639633),将CTG0070-PCR和VectorDNA2(CTG0009项目制备)连接;热转化至E.coliCompetentCellsJM109(CodeNo.9052)中,涂布平板,37℃过夜培养。使用T7/T7terminator引物挑选阳性菌落植菌,提取阳性克隆质粒CTG0123-2、3。使用T7、T7terminator、CTG0123P1、CTG0123P4引物对上述质粒进行测序。质粒CTG0123-2符合要求。3.蛋白表达及Westernblotting将质粒CTG0123-2转入BL21(DE3)T1R感受态细胞中,对阳性克隆进行诱导表达。3.1转化取质粒1μL转化competentcellBL21(DE3)T1R中,使用LB/抗生素Amp(100μg/ml)平板,35μL转化液涂布,37℃O/N培养,ControlpET28a(+)进行同样操作。3.2培养及诱导种培养:分别挑取单菌落至2mLLB/Amp(100μg/mL)培养基中,37℃O/N培养,命名为EXP0212-1。主培养诱导:分别在Glasstube中添加5mlLB/Amp(100μg/mL)培养基,添加种培养菌液100μL。37℃培养至OD600nm值约为0.6,添加100mMIPTG50μL(final1mMIPTG)进行诱导,37℃培养4h。pET28a诱导前吸光度值为0.60、集菌前吸光度值为1.15,EXP0212-1(CTG0123-2)诱导前吸光度值为0.60、集菌前吸光度值为2.45。蛋白质抽提:集菌后2.0OD相当的菌体加入320μLPBS悬浊后进行超声波破碎,对菌体破碎液进行离心分离(12000×rpm、5min)。取各抽提液(全蛋白、上清、沉淀)各8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min,使用12.5%polyacrylamidegel,电泳结束后,CBB-R250染色,使用脱色液脱色。结果见图15。3.3Westernblotting样品电泳对照取各抽提液(全蛋白、上清、沉淀)8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,使用彩色Marker及组氨酸标签Marker,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min,使用12.5%polyacrylamidegele,SDS-PAGE/CBB染色。将PVDF膜、滤纸分别剪切成与凝胶相同大小,使用转膜缓冲液处理后,按滤纸、PVDF膜、凝胶、滤纸的顺序依次放在转膜仪电极板之间,开始转膜,设置转膜仪参数如下:电流33mA,转印时间88min。将PVDF膜置于含1.5%BSA的10mLBlockingbuffer中,4℃平放过夜封闭。使用稀释后的Penta-HisAntibody溶液5mL,进行一次抗体反应1h。TBST缓冲液(20ml)洗涤2次;洗涤TBS缓冲液冲洗3次。使用稀释后的HRP-RabbitAnti-MouseIgG抗体溶液5mL,进行二次抗体反应1h。TBST缓冲液(20mL)洗涤2次;洗涤TBS缓冲液洗3次。1mLTrueBluePeroxidaseSubstrate显色1min。显色后PVDF膜如图16所示。结果表明EXP0212-1(CTG0123-2)为可溶性表达。4.EXP0212-1重组蛋白纯化及鉴定4.1500mL培养、表达与鉴定使用EXP0212-1甘油菌保进行500mL培养,以从菌体中进行目的蛋白纯化。具体步骤如下:(1)种培养:将EXP0212-1样品甘油菌保取50μL植入10mLLB/Kana(50μg/ml)培养基中,37℃O/N培养。(2)主培养及诱导:将10ml种培养菌液植入500mlLB/2LFlask中,37℃、200rpm培养至OD600≈0.6时,添加100mMIPTG500μL(final0.1mMIPTG)进行诱导,16℃继续培养24h。培养后离心,收集菌体。使用PBS清洗菌体后离心,称量菌体湿重。结果诱导前吸光度值为0.60,集菌前吸光度值为4.05,菌体湿重为2.72g。(3)SDS-PAGE电泳:取2.0OD相当的菌体加入320μLPBS悬浊后进行超声波破碎,对菌体破碎液进行离心分离(12000×rpm、5min),此即蛋白质抽提。取各抽提液(全蛋白、上清、沉淀)8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚、约80min,使用12.5%polyacrylamidegel,电泳结束后,CBB-R250染色,使用脱色液脱色,SDS-PAGE/CBB染色结果见图17,表明500mL培养菌体中有蛋白表达。5.2柱层析纯化与鉴定5.2.1缓冲液配制Sonicationbuffer/Washbuffer/BufferA,pH8.0(4℃),由50mMSodiumphosphate(pH8.0)、300mMNaCl、5mMImidazole组成;BufferB,pH8.0(4℃),由50mMSodiumphosphate(pH8.0)、300mMNaCl、300mMImidazole组成。透析Buffer为TaKaRaPBS(CodeNo.T900)。5.2.2菌体破碎培养后菌体湿重为2.72g,加入Sonicationbuffer(菌体湿重的10倍)30mL;4℃充分悬浊,Sonication180w,10min,收集破碎液30mL(S1);将S1置12000rpm、4℃离心30min、得到上清30mL(S2),沉淀0.99g;取S2使用0.45μm、1000mLVacuumfilter/Storagebottlesystem进行过滤,得到样品30mL。5.2.3柱层析纯化使用GEHiTrapTALONcrude,5mLTALONSuperflow(CodeNo.28-9537-66)柱层析和;使用10倍柱体积的灭菌MilliQH2O50mL和10倍柱体积的BufferA50mL平衡树脂,流速1mL/min。上样:BufferA平衡化后上样,流速为0.5mL/min。蛋白吸附后,使用10倍体积的BufferA50mL清洗树脂,流速1mL/min,W1:10mL,W2:20mL,W3:20mL。蛋白溶出Elution:BufferA50mL、BufferB50mL梯度溶出,流速1mL/min,Fraction1.1mL/tube×90tubes。5.2.4各阶段SDS-PAGE电泳及AKTA出峰图各纯化各阶段进行SDS-PAGE电泳,取全细胞(W)、上清(S)、不溶性沉淀(P)各0.05OD600电泳,取S1、S2、S3、Pa原液各1μL电泳,取W1、W2、W3原液各2.5μL电泳,取Elution原液各5μL电泳;取BSA标准品500ng上样。Proteinmarker:BroadWeightMarker(TaKaRaCode:3452Q),5μL/well。电泳胶为12.5%SDS-PAGE/考马斯亮蓝染色液染。5.2.5收集收集目的蛋白FractionNo.47~69,共计约26mL(S4)。取小量进行SDS-PAGE电泳并以BSA标准品浓度为参考。使用12.5%SDS-PAGE/考马斯亮蓝染色液染色。5.2.6透析使用PBS进行Buffer透析,反复进行3次,透析后样品为S5。透析Buffer为PBS,置换率为1:1000,透析膜:SnakeSkinDialysisTubing3500MWCO(CatNo.68035,LotNo.MH160055),使用12.5%SDS-PAGE/考马斯亮蓝染色液染色,取透析后样品进行SDS-PAGE电泳,如图18所示。使用Nanodrop-1000进行定量分析,目的蛋白的纯度:99%,目的蛋白总量:3.9mg,目的蛋白的浓度:0.15mg/mL。体积:26mL。9.Westernblotting检测取重组蛋白8μL(0.05OD相当),加入2μL5×SDSLoadingBuffer,95℃加热10min,进行SDS-PAGE电泳。电泳条件:25mA/枚,70min。使用12.5%polyacrylamidegel,电泳结束后,CBB-250染色,使用脱色液脱色。将PVDF膜、滤纸分别剪切成与凝胶相同大小,使用转膜缓冲液处理后,按滤纸、PVDF膜、凝胶、滤纸的顺序依次放在转膜仪电极板之间,2张膜同时开始转膜,设置转膜仪参数如下:电流52mA、转印时间80min。将PVDF膜置于含1.5%BSA的12mLBlockingbuffer中,37℃平放1h封闭。使用1:10稀释后的血清溶液10mL,进行一次抗体反应1h。TBST缓冲液(25mL)洗涤1次;洗涤TBS缓冲液冲洗2次。使用1:2500稀释后的GoatAnti-humanIgE(HRP)抗体溶液9mL进行二次抗体反应1h。TBST缓冲液(25mL)洗涤1次;洗涤TBS缓冲液冲洗2次。1mLTrueBluePeroxidasesubstrate显色1min。显色后PVDF膜如图19所示。实施例4:以重组蛋白为抗原,Westernblotting检测过敏性疾病哮喘/鼻炎患者血清特异性IgE,诊断患者是否对该重组蛋白过敏。通过临床实验验证本发明所制备的纯化蛋白在临床中的应用,应用结果如下表:SpecificIgE-bindingtorecombinantallergensThereactivitiesofthespecificIgEantibodiestorecombinantallergenswereexaminedbyWesternblotting.Inbrief,8μLoftherecombinantallergenswereelectrophoresedona12.5%polyacrylamidegelandelectroblottedontopolyvinylidenedifluoride(PVDF,Millipore,CodeNo.IPVH20200)membrane.Themembranewasblockedwith1.5%bovineserumalbuminin1×TBS-Tfor1hatroomtemperature.Afterovernightincubationwith1:10dilutionoftheT.putrescentiae-positivehumanserainblockingbufferat4℃,themembranewasincubatedwith1:2500dilutionofGoatanti-humanIgE-HRP(secondaryantibody,Invitrogen,Carlsbad,Calif.,USA)for1h.Themembranewaswashed1timeswithTBS-TandTBSbuffer2times.1mLofTrueBluePeroxidasesubstratewasaddedtothemembranesforcoloration;2minafterreaction,thefilmwasexposedandimaged.以上所述实施例只是本发明的较佳实例,而没有限制本发明的实施范围。因此,凡是根据本
发明内容的实质所作出的等效修改,都应属于在本发明的保护范围内。<120>Title:<130>AppFileReference:<140>CurrentAppNumber:<141>CurrentFilingDate:Sequence--------<213>OrganismName:<400>PreSequenceString:aatgggtcgcggatccatgtcccaagtaccgatcaa36<212>Type:DNA<211>Length:36SequenceName:1SequenceDescription:Sequence--------<213>OrganismName:<400>PreSequenceString:atctcagtggtggtggtggtggtgctcgagtgcggccgcttactgcccgccagcaagct59<212>Type:DNA<211>Length:59SequenceName:2SequenceDescription:Sequence--------<213>OrganismName:<400>PreSequenceString:cactgatgctcgcctggaag20<212>Type:DNA<211>Length:20SequenceName:3SequenceDescription:Sequence--------<213>OrganismName:<400>PreSequenceString:AATGGGTCGCGGATCCATGTCTTGGCAATCCTACGT36<212>Type:DNA<211>Length:36SequenceName:4SequenceDescription:Sequence--------<213>OrganismName:<400>PreSequenceString:ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGTCAATAGTTGTTGGCTTTCA50<212>Type:DNA<211>Length:50SequenceName:5SequenceDescription:Sequence--------<213>OrganismName:<400>PreSequenceString:AATGGGTCGCGGATCCATGGCCAACAAGAAGACGCC36<212>Type:DNA<211>Length:36SequenceName:6SequenceDescription:Sequence--------<213>OrganismName:<400>PreSequenceString:ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGTGCGGCCGCCTAGTCCACTTCCTCAATGG59<212>Type:DNA<211>Length:59SequenceName:7SequenceDescription:当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1