一种ALS患者特异性运动神经元的制备方法与流程
本发明属于生物技术领域,具体一种ALS患者特异性运动神经元的制备方法。
背景技术:
人肌肉萎缩侧索硬化症(Amyotrophic lateral sclerosis,简称ALS)是一种神经退行性疾病,主要病理特征为运动神经元退行性病变,导致病人肌肉萎缩,瘫痪,甚至会影响呼吸系统,危及病人生命。ALS发病迅速,多数病人发病3-5年后死亡。ALS分为两种类型,散发性(sporadic ALS,SALS)和家族性(familial ALS,FALS)。二者临床症状类似,但是致病机理都不清楚,也没有有效的药物治疗。SALS成因复杂,可能由于环境因素和遗传因素共同作用。研究发现FALS一般是由某基因杂合突变引起,如SOD1,FUS,C9ORF72,TARDBP等。SOD1基因突变是最早发现与FALS相关的致病基因,由SOD1基因突变导致的ALS病人约占FALS病人的20%。针对SOD1基因突变的致病机理研究大多集中在模式生物(如小鼠)中进行。研究发现,SOD1基因突变影响生物体的氧化还原稳态,容易形成疏水的凝聚物,干扰蛋白质降解途径,破环蛋白质稳态,影响线粒体功能,引起细胞凋亡。最近还有报道称,SOD1作为转录因子调控细胞的氧化应激反应。FUS是RNA结合蛋白,参与调控mRNA的编辑,转运和翻译过程。关于FUS的详细机制研究相对较少,在ALS病人的病理检查中发现异常的FUS凝聚物。
虽然在模式生物(如小鼠)中研究ALS有助于我们了解ALS的机制。但是利用模式生物筛选有效的药物和探索其他有效地治疗方案的进展缓慢。迄今为止,只有一种治疗ALS的药物利鲁唑被美国食品药物管理局许可,但其药效缓慢。原因之一是种属差异性,由于人和模式生物的遗传背景不同,在模式生物中筛选的有效治疗ALS的药物在临床上不能缓解ALS症状,或者副作用大。其次,通过过表达突变基因产生的ALS疾病模型不能真正反映病人的生理状况。外源过表达突变致病基因的表达水平远远高于病人体内单位点基因突变的表达水平,从而掩盖了某些致病的关键因素,不能真正反应致病机理,在此基础上筛选的药物也不能有效缓解ALS病人的临床症状。
技术实现要素:
本发明的目的是提供一种人肌肉萎缩侧索硬化症病人特异性运动神经元的制备方法。
本发明所提供的人肌肉萎缩侧索硬化症病人特异性运动神经元的制备方法,可为如下方法A或方法B:
方法A:不携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元的制备方法,具体可包括如下步骤:
(1)对离体的携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人来源的成纤维细胞进行重编程,得到携带所述ALS相关基因突变的诱导多能干细胞,记为ALS-iPSC;
(2)对所述ALS-iPSC进行基因编辑,特异性消除所述ALS相关基因突变,得到基因矫正的诱导多能干细胞,记为cALS-iPSC;
(3)对所述cALS-iPSC进行定向诱导分化,即得不携带所述ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元。
方法B:携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元的制备方法,具体可包括如下步骤:
(1)对离体的携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人来源的成纤维细胞进行重编程,得到携带所述ALS相关基因突变的诱导多能干细胞,记为ALS-iPSC;
(2)对所述ALS-iPSC进行定向诱导分化,即得携带所述ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元。
其中,所述重编程指不改变基因序列的情况下,通过表观遗传修饰(如DNA甲基化)来改变细胞命运的过程。目前重编程主要指两个过程:其一,分化的细胞逆转恢复到全能性状态的过程;其二,从一种分化细胞转化为另一种分化细胞的过程。
在所述方法的步骤(1)中,所述重编程具体为将OCT3/4基因、SOX2基因、KLF4基因、LMyc基因和LIN28基因共同导入(如电转)所述离体的携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人来源的成纤维细胞中,并培养至获得所述ALS相关基因突变的诱导多能干细胞。
在本发明中,所述“将OCT3/4基因、SOX2基因、KLF4基因、L-MYC基因和LIN28基因共同导入所述离体的携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人来源的成纤维细胞中”具体是通过将携带有所述OCT3/4基因的pCXLE-hOCT3/4-shp53-F质粒、携带有所述SOX2基因和所述KLF4基因的pCXLE-hSK质粒和携带有所述L-MYC基因和所述LIN28基因的pCXLE-hUL质粒共同导入(如电转)所述离体的携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人来源的成纤维细胞中实现的。在实际操作中,为了便于阳性克隆的筛选,可以同时导入pCXLE-EGFP质粒该质粒携带并表达EGFP基因。四种质粒的质量配比可为1:1:1:1。
步骤(1)中,所述培养具体可为先在成纤维细胞培养基中培养1天后换液,用所述成纤维细胞培养基继续培养至第5天后转接到含有预先用丝裂霉素灭活处理过的小鼠胚胎成纤维细胞的饲养层体系中用iPSC培养基培养至第3周,获得所述ALS相关基因突变的诱导多能干细胞。
其中,所述成纤维细胞培养基的组成如下:将DMEM培养基(Invitrogen,货号为11965118)和FBS(Hyclone)按照体积比为9:1的比例混合后,加入青霉素/链霉素(Invitrogen,货号为15070-063),使所述青霉素/链霉素的终浓度为10g/L。
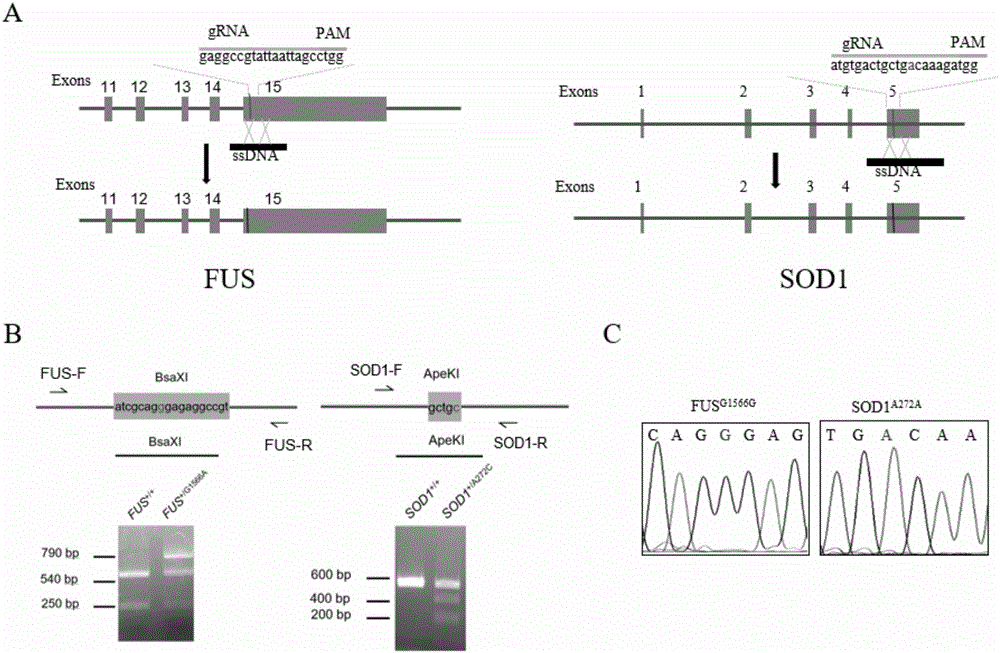
在本发明中,所述ALS相关基因突变具体可为FUS G1566A或SOD1 A272C。
所述FUS G1566A是指FUS基因(基因组序列为GenBank:NG_012889.2,cDNA序列为GenBank:NM_004960.3)的基因编码序列(CDS序列)的第1566位发生了从G到A的突变,突变后的基因可简称为FUSG1566A。所述SOD1 A272C是指SOD1基因(基因组序列为GenBank:NG_008689.1,cDNA序列为GenBank:NM_000454.4)的基因编码序列(CDS序列)的第272位发生了从A到C的突变,突变后的基因可简称为SOD1A272C。
相应的,步骤(1)中,所述离体的携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人来源的成纤维细胞可为皮肤来源的成纤维细胞,具体可为FUS G1566A成纤维细胞或者SOD1 A272C成纤维细胞;所述FUS G1566A成纤维细胞具体为来自于美国Coriell Cell Repository的parent cell line为ND29563的细胞;所述SOD1 A272C成纤维细胞具体为来自于美国Coriell Cell Repository的parent cell line为ND29149的细胞。
所述方法A的步骤(2)中,所述“特异性消除所述ALS相关基因突变”为将所述FUS G1566A矫正为FUS G1566G,或将所述SOD1 A272C矫正为SOD1 A272A。
在本发明中,所述方法A的步骤(2)中,对所述ALS-iPSC进行基因编辑具体为利用CRISPR/Cas9技术对所述ALS-iPSC进行基因编辑。
相应的,所述方法A的步骤(2)具体为:将gRNA表达质粒、Cas9表达质粒和重组模板共同转入所述ALS-iPSC,并培养至获得所述cALS-iPSC。
当所述ALS相关基因突变为所述FUS G1566A时,所述gRNA表达质粒中的gRNA的识别区域序列(即间隔序列spacer对应的编码序列)为序列表中序列1;所述重组模板为序列表中序列2所示的双链DNA;所述gRNA表达质粒具体为FUS-gRNA-mCherry;所述FUS-gRNA-mCherry为将序列表中序列1所示的DNA片段插入到gRNA-mCherry载体(Addgene,货号为#78544)的酶切位点AflII间后得到的重组质粒。
当所述ALS相关基因突变为所述SOD1 A272C时,所述gRNA表达质粒中的gRNA的识别区域序列(即间隔序列spacer对应的编码序列)为序列表中序列3;所述重组模板为序列表中序列4所示的双链DNA。所述gRNA表达质粒具体为SOD1-gRNA-mCherry;所述SOD1-gRNA-mCherry为将序列表中序列2所示的DNA片段插入到gRNA-mCherry载体(Addgene,货号为#78544)的酶切位点AflII间后得到的重组质粒。
所述Cas9表达质粒为Cas9-GFP;所述Cas9-GFP购自美国Addgene公司,货号为#59766。
其中,所述gRNA表达质粒、所述Cas9表达质粒和所述重组模板的质量配比为1:2:2。
所述方法A的步骤(2)中,所述培养具体为将成功转入所述gRNA表达质粒、所述Cas9表达质粒和所述重组模板的所述ALS-iPSC转接到含有预先用丝裂霉素灭活处理过的小鼠胚胎成纤维细胞的饲养层体系中用cDF12培养基(具体配方见下文)培养2周,获得所述cALS-iPSC。
对所述cALS-iPSC进行阳性鉴定的方法具体可为CAPs鉴定(用PCR技术扩增出包含突变位点的片段,由于一个SNP的差异,可以用相应的限制性内切酶鉴定)。最终得到把突变基因FUSG1566A矫正为FUSG1566G的克隆,突变基因SOD1A272C矫正为SOD1A272A的克隆,即为阳性克隆。
FUS-F:5’-GAGAAAGTGGTTTCATTTTGAGGGCTAGGTGGA-3’;
FUS-R:5’-TTGTTTGAGCCTCACCATTAAAAGGGCCAAAAG-3’。
FUS鉴定用限制性内切酶:BSAXI。
SOD1-F:5’-CCCATCTTTCTTCCCAGAGCATTAGTGTGTAGACG-3’;
SOD1-R:5’-ACAAAATGTTCTGTTTAACAAGTGAGAAACCCAATCCT-3’。
SOD1鉴定用限制性内切酶:APEKI。
在所述方法A的步骤(3)中或所述方法B的步骤(2)中,对所述cALS-iPSC或所述ALS-iPSC进行定向诱导分化具体是按照包括如下步骤的方法进行的:
(3-1)将所述cALS-iPSC或所述ALS-iPSC接种到含有预先用丝裂霉素灭活处理过的小鼠胚胎成纤维细胞的饲养层体系中用cDF12培养基培养,得到待分化细胞;
(3-2)在MN培养基1中对所述待分化细胞进行第一次分化培养,得到第一次培养后细胞;
(3-3)在MN培养基2中对所述第一次培养后细胞进行第二次分化培养,得到第二次培养后细胞;
(3-4)在MN培养基3中对所述第二次培养后细胞进行第三次分化培养,得到第三次培养后细胞;
(3-5)在MN培养基4中对所述第三次培养后细胞进行第四次分化培养,得到第四次培养后细胞;
(3-6)在MN培养基5中对所述第四次培养后细胞进行第五次分化培养,得到第五次培养后细胞,即为不携带所述ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元或者携带所述ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元。
其中,步骤(3-5)中,进行所述第四次分化培养时,是将所述第三次培养后细胞以单细胞转到经基质胶(Matrigel)包被的培养板中用所述MN培养基4进行培养的。在该步骤中,还可于传代前后用rock inhibitor处理以提高细胞存活率。
其中,所述cDF12培养基的组成如下:将DMEM/F12培养基(Invitrogen,货号为11320-033)和Knockout血清替代物(Invitrogen,货号为N10828-028)按照8:2的体积比混合后,向混合液中加入非必需氨基酸(Invitrogen,货号为11140-050)、GlutaMAX(Invitrogen,货号为35050-061)、青霉素/链霉素(Invitrogen,货号为15070-063)、β-巯基乙醇(Invitrogen,21985-023)和人FGF2(Joint Protein Central),使得所述非必需氨基酸的终浓度为0.1mM,所述GlutaMAX的终浓度为1mM,所述青霉素/链霉素的终浓度为10g/L,所述β-巯基乙醇的终浓度为55μM,所述人FGF2的终浓度为10ng/ml。
所述MN培养基1的组成如下:将Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和Neurobasal培养基(Invitrogen,货号为12348017)按照1:1的体积比混合后,向混合液中加入N2(Invitrogen,货号为17502048)、B27(Invitrogen,货号为17504044)、GlutaMAX(Invitrogen,货号为35050-061)、肝素(Heparin)(Sigma,货号为H3149)、CHIR99021(Cellagentech,货号为C2447)、SB431542(Cell agentech,货号为C7243)、Compound E(EMD Chemicals,货号为209986-17-4)和Dorsomorphin(Sigma,货号为P5499);使得所述N2的终浓度为1%(体积百分含量),所述B27的终浓度为2%(体积百分含量),所述GlutaMAX的终浓度为2mM,所述肝素(Heparin)的终浓度为2g/L,所述CHIR99021的终浓度为4μM,所述SB431542的终浓度为3μM,所述Compound E的终浓度为0.1μM,所述Dorsomorphin的终浓度为1μM。
所述MN培养基2的组成如下:将Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和Neurobasal培养基(Invitrogen,货号为12348017)按照1:1的体积比混合后,向混合液中加入N2(Invitrogen,货号为17502048)、B27(Invitrogen,货号为17504044)、GlutaMAX(Invitrogen,货号为35050-061)、肝素(Heparin)(Sigma,货号为H3149)、CHIR99021(Cellagentech,货号为C2447)、SB431542(Cell agentech,货号为C7243)、Compound E(EMD Chemicals,货号为209986-17-4)、Dorsomorphin(Sigma,货号为P5499)和RA(Sigma,货号为R2625);使得所述N2的终浓度为1%(体积百分含量),所述B27的终浓度为2%(体积百分含量),所述GlutaMAX的终浓度为2mM,所述肝素(Heparin)的终浓度为2g/L,所述CHIR99021的终浓度为4μM,所述SB431542的终浓度为3μM,所述Compound E的终浓度为0.1μM,所述Dorsomorphin的终浓度为1μM,所述RA的终浓度为100nM。
所述MN培养基3的组成如下:将Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和Neurobasal培养基(Invitrogen,货号为12348017)按照1:1的体积比混合后,向混合液中加入N2(Invitrogen,货号为17502048)、B27(Invitrogen,货号为17504044)、GlutaMAX(Invitrogen,货号为35050-061)、肝素(Heparin)(Sigma,货号为H3149)、CHIR99021(Cellagentech,货号为C2447)、SB431542(Cell agentech,货号为C7243)、Compound E(EMD Chemicals,货号为209986-17-4)、RA(Sigma,货号为R2625)和SAG(货号为364590-63-6);使得所述N2的终浓度为1%(体积百分含量),所述B27的终浓度为2%(体积百分含量),所述GlutaMAX的终浓度为2mM,所述肝素(Heparin)的终浓度为2g/L,所述CHIR99021的终浓度为4μM,所述SB431542的终浓度为3μM,所述Compound E的终浓度为0.1μM,所述RA的终浓度为100nM,所述SAG的终浓度为500ng/ml。
所述MN培养基4的组成如下:将Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和Neurobasal培养基(Invitrogen,货号为12348017)按照1:1的体积比混合后,向混合液中加入N2(Invitrogen,货号为17502048)、B27(Invitrogen,货号为17504044)、GlutaMAX(Invitrogen,货号为35050-061)、肝素(Heparin)(Sigma,货号为H3149)、Compound E(EMD Chemicals,货号为209986-17-4)、RA(Sigma,货号为R2625)和SAG(货号为364590-63-6);使得所述N2的终浓度为1%(体积百分含量),所述B27的终浓度为2%(体积百分含量),所述GlutaMAX的终浓度为2mM,所述肝素(Heparin)的终浓度为2g/L,所述Compound E的终浓度为0.1μM,所述RA的终浓度为100nM,所述SAG的终浓度为500ng/ml。
所述MN培养基5的组成如下:将Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和Neurobasal培养基(Invitrogen,货号为12348017)按照1:1的体积比混合后,向混合液中加入N2(Invitrogen,货号为17502048)、B27(Invitrogen,货号为17504044)、GlutaMAX(Invitrogen,货号为35050-061)、肝素(Heparin)(Sigma,货号为H3149)、Compound E(EMD Chemicals,货号为209986-17-4)、DAPT(Sigma,货号为D5942)和纤维连接蛋白(fibronectin)(Gibco,货号为PHE0023);使得所述N2的终浓度为1%(体积百分含量),所述B27的终浓度为2%(体积百分含量),所述GlutaMAX的终浓度为2mM,所述肝素(Heparin)的终浓度为2g/L,所述Compound E的终浓度为0.1μM,所述DAPT的终浓度为10μM,所述纤维连接蛋白(fibronectin)的终浓度为10μg/ml。
在步骤(3-1)中,所述培养的时间具体可为1-2天。在步骤(3-2)中,所述培养的时间为3天。在步骤(3-3)中,所述培养的时间具体可为3天。在步骤(3-4)中,所述培养的时间具体可为1天。在步骤(3-5)中,所述培养的时间具体可为1天。在步骤(3-6)中,所述培养的时间具体可为3天。
本发明还请求保护一种用于制备人肌肉萎缩侧索硬化症病人特异性运动神经元的试剂盒。
本发明所要求保护的用于制备人肌肉萎缩侧索硬化症病人特异性运动神经元的试剂盒,含有所述MN培养基1、所述MN培养基2、所述MN培养基3、所述MN培养基4、所述MN培养基5。
当然也可以含有如上所述的cDF12培养基。
利用所述方法制备得到的人肌肉萎缩侧索硬化症病人特异性运动神经元也属于本发明的保护范围。
所述方法或所述试剂盒或所述人肌肉萎缩侧索硬化症病人特异性运动神经元在如下任一中的应用也属于本发明的保护范围:
(A)制备用于筛选和/或鉴定能够治疗和/或预防人肌肉萎缩侧索硬化症的临床药物和/或天然有机物和/或小分子化合物的产品(主要为携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元的应用);
(B)制备用于治疗和/或预防人肌肉萎缩侧索硬化症的产品(主要为不携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元的应用);
(C)制备人肌肉萎缩侧索硬化症的细胞模型(主要为携带ALS相关基因突变的人肌肉萎缩侧索硬化症病人特异性运动神经元的应用)。
本发明制备的ALS特异性运动神经元具有如下优点:1)在体外具有自我更新和分化成三个胚层的潜能,因而可以定向分化成受疾病累积的细胞系;2)利用现有的基因打靶技术可以针对病人自体来源的诱导型多能干细胞进行基因突变的基因组原位矫正,从而在基因水平清除内源性致病因素;3)病人自体来源的iPSC,可以进一步分化为可供自身移植的细胞,避免了配型和排斥反应的困扰。
本发明利用iPSC技术将ALS病人来源的皮肤成纤维细胞,通过非整合附加型质粒电转的方法使其重编程为其特异性的iPSC,利用CRISPR/Cas9基因编辑技术靶向矫正基因突变,得到遗传背景一致(isogenic)的对照,并且同时定向分化成运动神经元。本发明获得的携带ALS致病基因突变的运动神经元可以作为有效的平台进行高效高通量的个性化药物筛选,不携带ALS致病基因突变的运动神经元有望用于ALS的治疗与预防。本发明为ALS疾病研究、开发疾病模型、研究疾病的发病机制并进行疾病治疗奠定基础,在个性化治疗和转化医学中具有巨大的应用前景。
附图说明
图1为两株ALS病人来源的皮肤成纤维细胞的重编程和全能性鉴定。其中,A为ALS病人来源的皮肤成纤维细胞(FUS G1566A成纤维细胞和SOD1 A272C成纤维细胞)和重编程得到的对应iPSC的形态。B为ALS病人来源的细胞所携带的基因突变。C为免疫荧光检测干细胞标记物NANOG,OCT4,SOX2。D为将ALS病人特异的iPSC打进免疫缺陷小鼠的皮下,分化成具有三个胚层的畸胎瘤,其中,TUJ1(外胚层标记物),FOXA2(内胚层标记物),α-SMA(中胚层标记物)。
图2为靶向矫正ALS病人携带的基因突变。其中,A为基因矫正方案示意图;B为CAPs鉴定消除基因突变的结果;C为测序确认消除基因突变。
图3为将所述iPSC定向分化成运动神经元细胞。其中,A为将iPSC定向分化成运动神经元细胞的方案示意图;B为FUSG1566A-MN,SOD1A272C-MN,FUSG1566G-MN,SOD1A272A-MN运动神经元细胞形态;C为免疫荧光检测运动神经元细胞的标记物HB9和ISL1,以及神经元细胞的标记物MAP2。
图4为携带ALS致病基因突变的运动神经元和经基因矫正的运动神经元之间差异基因表达的转录组分析结果。其中,A为FUSG1566A-MN/FUSG1566G-MN组和SOD1A272C-MN/SOD1A272A-MN组的上调和下调基因分析;B为FUSG1566A-MN/FUSG1566G-MN组和SOD1A272C-MN/SOD1A272A-MN组组内上调和下调基因分析;C为FUSG1566A-MN/FUSG1566G-MN组和SOD1A272C-MN/SOD1A272A-MN组两组共同上调和下调基因分析。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
下述实施例中的细胞培养条件如无特殊说明,均为37℃,5%CO2。
下述实施例中的FUS G1566A成纤维细胞(ND29563)、SOD1 A272C成纤维细胞(ND29149)均购买于美国的Coriell Cell Repository。
下述实施例中的pCXLE-hOCT3/4-shp53-F(#27077)、pCXLE-hSK(#27078)、pCXLE-hUL(#27080)和pCXLE-EGFP(#27082)均购买于Addgene。其中,pCXLE-hOCT3/4-shp53-F携带并表达OCT3/4基因;pCXLE-hSK携带并表达SOX2基因和KLF4基因;pCXLE-hUL携带并表达L-MYC基因和LIN28基因;pCXLE-EGFP携带并表达EGFP基因。
下述实施例中的用于免疫荧光的抗体如下:
抗人OCT4抗体(sc-5279),Santa Cruz Biotechnology。
抗人SOX2抗体(sc-17320),Santa Cruz Biotechnology。
抗人NANOG抗体(ab21624),Abcam。
抗人TUJ1抗体(T2220),Sigma。
抗人FOXA2抗体(8186),Cell Signaling Technology。
抗人SMA抗体(A5228),Sigma。
抗人MAP2抗体(4403),Sigma。
抗人HB9抗体(81.5C10),Developmental Studies Hybridoma Bank
抗人ISL1抗体(ab20670),Abcam。
下述实施例中的培养基配方如下:
(1)成纤维细胞培养基:将90%(体积百分含量)DMEM培养基(Invitrogen,货号为11965118)和10%(体积百分含量)FBS(Hyclone,货号为SH30084.03)混合后,加入如下物质:1%(10g/L)青霉素/链霉素(Invitrogen,货号为15070-063),其中各物质的浓度为在成纤维细胞培养基中的终浓度。
(2)iPSC培养基配方(cDF12培养基):将80%(体积分数)DMEM/F12培养基(Invitrogen,货号为11320-033)和20%(体积百分含量)Knockout血清替代物(Invitrogen,货号为N10828-028)混合后,加入如下物质:0.1mM非必需氨基酸(Invitrogen,货号为11140-050)、1mM GlutaMAX(Invitrogen,货号为35050-061)、1%(10g/L)青霉素/链霉素(Invitrogen,货号为15070-063)、55μMβ-巯基乙醇(Invitrogen,货号为21985-023)、10ng/ml Human FGF2(Joint Protein Central),其中各物质的浓度为在iPSC培养基中的终浓度。
(3)MN培养基1配方:将50%(体积百分含量)Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和50%(体积百分含量)Neurobasal培养基(Invitroge,货号为12348017)混合后,加入如下物质:1%(体积百分含量)N2(Invitrogen,货号为17502048)、2%(体积百分含量)B27(Invitrogen,货号为17504044)、2mM GlutaMAX(Invitrogen,货号为35050-061)、0.2%(2g/L)Heparin(Sigma,货号为H3149)、4μM CHIR99021(Cell agentech,货号为C2447)、3μM SB431542(Cellagentech,货号为C7243)、0.1μM Compound E(EMD Chemicals,货号为209986-17-4)、1μM Dorsomorphin(Sigma,货号为P5499),其中各物质的浓度为在MN培养基1中的终浓度。
(4)MN培养基2配方:将50%(体积百分含量)Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和50%(体积百分含量)Neurobasal培养基(Invitroge,货号为12348017)混合后,加入如下物质:1%(体积百分含量)N2(Invitrogen,货号为17502048)、2%(体积百分含量)B27(Invitrogen,货号为17504044)、2mM GlutaMAX(Invitrogen,货号为35050-061)、02%(2g/L)Heparin(Sigma,货号为H3149)、4μM CHIR99021(Cell agentech,货号为C2447)、3μM SB431542(Cellagentech,货号为C7243)、0.1μM Compound E(EMD Chemicals,货号为209986-17-4)、1μM Dorsomorphin(Sigma,货号为P5499)、100nM Retinoic Acid(Sigma,货号为R2625),其中各物质的浓度为在MN培养基2中的终浓度。
(5)MN培养基3配方:将50%(体积百分含量)Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和50%(体积百分含量)Neurobasal培养基(Invitroge,货号为12348017)混合后,加入如下物质:1%(体积百分含量)N2(Invitrogen,货号为17502048)、2%(体积百分含量)B27(Invitrogen,货号为17504044)、2mM GlutaMAX(Invitrogen,货号为35050-061)、0.2%(2g/L)Heparin(Sigma,货号为H3149)、4μM CHIR99021(Cellagentech,货号为C2447)、3μM SB431542(Cellagentech,货号为C7243)、0.1μM Compound E(EMD Chemicals,货号为209986-17-4)、100nM Retinoic Acid(Sigma,货号为R2625)、500ng/ml SAG(Calbiochem,货号为364590-63-6),其中各物质的浓度为在MN培养基3中的终浓度。
(6)MN培养基4配方:将50%(体积百分含量)Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和50%(体积百分含量)Neurobasal培养基(Invitroge,货号为12348017)混合后,加入如下物质:1%(体积百分含量)N2(Invitrogen,货号为17502048)、2%(体积百分含量)B27(Invitrogen,货号为17504044)、2mM GlutaMAX(Invitrogen,货号为35050-061)、0.2%(2g/L)Heparin(Sigma,货号为H3149)、0.1μM Compound E(EMD Chemicals,货号为209986-17-4)、100nM Retinoic Acid(Sigma,货号为R2625)、500ng/ml SAG(货号为364590-63-6),其中各物质的浓度为在MN培养基4中的终浓度。
(7)MN培养基5配方:将50%(体积百分含量)Advanced DMEM/F12培养基(Invitrogen,货号为12634028)和50%(体积百分含量)Neurobasal培养基(Invitroge,货号为12348017)混合后,加入如下物质:1%(体积百分含量)N2(Invitrogen,货号为17502048)、2%(体积百分含量)B27(Invitrogen,货号为17504044)、2mM GlutaMAX(Invitrogen,货号为35050-061)、0.2%(2g/L)Heparin(Sigma,货号为H3149)、0.1μM Compound E(EMD Chemicals,货号为209986-17-4)、10μM DAPT(Sigma,货号为D5942)、10μg/ml fibronectin(Gibco,货号为PHE0023),其中各物质的浓度为在MN培养基5中的终浓度。
下述实施例中的细胞培养方法如下:
(1)成纤维细胞的培养
用所述成纤维细胞培养基培养,隔天换液,每3-4天传一次代。传代时按1:3或1:4传代。
(2)iPSC的培养
(2-1)饲养层体系:将iPSC接种至预先铺好的经过丝裂霉素(美国Sigma公司产品,货号:M0503)灭活处理的小鼠胚胎成纤维细胞(简称MEF,美国Invitrogen公司产品,货号:S1520-100)的培养板中,与MEF共同培养,使用所述cDF12培养基培养,每5-7天传一次代。
(2-2)无饲养层体系:将iPSC接种至预先用细胞外基质(Growth factor reduced-Matrigel,美国BD Biosciences产品,货号:354277)包被的培养板中,使用mTeSR培养基(美国StemCell Technologies产品,货号:#05850)培养,每5-7天传一次代。
(3)运动神经元的培养
将定向诱导分化的运动神经元前体细胞接种至预先用matrigel包被的培养板中,用MN培养基5培养。
实施例1、获得ALS患者来源的诱导多能干细胞(iPSC)
一、ALS患者来源的诱导多能干细胞的获得
1、细胞培养
分别将两株携带不同基因突变的ALS病人来源的皮肤成纤维细胞:FUS G1566A成纤维细胞(ND29563)和SOD1 A272C成纤维细胞(ND29149)在成纤维细胞培养基中进行扩大培养。待细胞培养到合适密度,消化,计数,两种成纤维细胞各取1.5×106个。
2、电转液制备
将100μl opti-MEM(life technology产品)、1.5μg pCXLE-hOCT3/4-shp53-F、1.5μg pCXLE-hSK、1.5μg pCXLE-hUL和1.5μg pCXLE-EGFP混匀,得到电转液,置于1.5ml EP管中。
3、用步骤2获得的电转液分别对步骤1获得的FUS G1566A成纤维细胞(ND29563)和SOD1 A272C成纤维细胞(ND29149)分别进行重悬,重悬后转移到电转杯里,并放入电转仪(Lonza 4D nucleofector)中,进行电转,电转程序选择EN150(具体步骤参考Lonza 4D nucleofector说明书),分别得到电转后的细胞悬液(1.5×106细胞/100μl)。
4、分别将电转后的细胞悬液加入到预热的含有成纤维细胞培养基的六孔板的孔中,摇晃均匀,放回培养箱中培养。第二天换液,观察电转效率。
5、持续用成纤维细胞培养基培养至第5天,消化接种至预先铺好的经过丝裂霉素灭活处理的MEF的培养板中,使用iPSC培养基持续培养。培养至第三周,分别得到类似于胚胎干细胞的小克隆(iPSC),将FUS G1566A成纤维细胞(ND29563)诱导得到的iPSC命名为FUSG1566A突变的iPSC(简称为FUSG1566A-iPSC),将SOD1 A272C成纤维细胞(ND29149)诱导得到的iPSC命名为SOD1A272C突变的iPSC(简称为SOD1A272C-iPSC),不同成纤维细胞诱导得到的iPSC的形态如图1中A所示,基因突变结果如图1中B所示。分别将小克隆挑至预先铺好的经过丝裂霉素灭活处理的MEF的培养板中扩大培养。
二、ALS患者来源的诱导多能干细胞的鉴定
通过在体外检测干细胞标记物和体内检测向三胚层形成能力(畸胎瘤实验)表明,所述iPSC具有与胚胎干细胞类似的干性。具体如下:
1、免疫荧光检测干细胞标记物NANOG,OCT4,SOX2的表达
分别将步骤一所得的两种携带基因突变的iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC)接种到预先铺好经过丝裂霉素灭活处理的MEF的盖玻片上,待其贴壁后,先用4%多聚甲醛固定30分钟后,再用磷酸盐缓冲液(PBS)润洗3次,再用含0.4%(体积百分含量)TritonX-100的PBS通透30分钟,PBS洗3次。然后用10%(体积百分含量)的驴血清封闭1个小时。一抗按照适当比例(抗人鼠源OCT4抗体,sc-5279,1:100;抗人羊源SOX2抗体,sc-17320,1:100;抗人兔源NANOG抗体,ab21624,1:250)稀释后于4℃孵育过夜。第二天,再用PBS洗3次后,加入适当比例稀释的二抗和细胞核染料Hoechst33342(Invitrogen,货号:H3569)孵育1个小时。PBS洗3次后,用封片剂(Vector公司,货号H-1000)封片。最后在激光共聚焦显微镜下进行观察,拍照。
检测结果如图1中C所示:从图中可以看出,获得的两种携带基因突变的iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC)均表达OCT4,NANOG,SOX2这三个干细胞的标记物,说明两种携带基因突变的iPSC均保持有很好的干性。
2、畸胎瘤实验
分别将步骤一所得的两种携带基因突变的iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC)接种到预先铺好经过丝裂霉素灭活处理的MEF的培养板中。待iPSC长满,消化计数,并用含20%(体积百分含量)Matrigel和80%(体积百分含量)cDF12培养基的混合液对iPSC(3×106个)进行重悬,得到细胞悬液;并将获得的细胞悬液打入免疫缺陷小鼠皮下。大约8周后,即有黄豆大小的畸胎瘤长成。脱颈处死小鼠后,将畸胎瘤取出,于4%多聚甲醛中固定一周,使其充分固定后,泡在30%(30g/100ml)蔗糖溶液中脱水至畸胎瘤沉底,表明已充分脱水。然后,使用Compound O.C.T包埋剂进行包埋。在冰冻切片机上进行切片,厚度为12μm,组织切片晾干后,即可进行免疫荧光检测三个胚层标记物的表达。免疫荧光操作步骤同上述步骤1。其中,一抗包括:抗人兔源TUJ1抗体(Sigma公司,货号T2220,1:500);抗人兔源FOXA2抗体(Cell Signaling Technology公司,货号8186,1:200);抗人鼠源-SMA抗体(Sigma公司,货号A5228,1:200)。
检测结果如图1中D所示:可见由两种携带基因突变的iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC)形成的畸胎瘤均表达TUJ1,FOXA2,SMA这三个胚层的标记物,说明两种携带基因突变的iPSC均保持有很好的干性。
实施例2、靶向矫正两种ALS-iPSC携带的基因突变
利用CRISPR/Cas9基因编辑技术对实施例1获得的两种iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC)携带的基因突变进行靶向清除,最终得到消除基因突变的iPSC。具体步骤如下:
1、细胞培养
在基质胶(matrigel)包被的培养板中培养所述实施例1获得的两种iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC),培养基为mTESR。待克隆密度达到70%-80%,TrypLE消化,计数,收集5×106个细胞。
2、电转液制备
将8μg Cas9-GFP质粒,8μg相应基因矫正的重组模板和4μg相应基因矫正的gRNA-mCherry质粒混匀,用opti-MEM(life technology)定容至100μl,得到电转液,置于1.5ml EP管中。
当需要矫正的相关基因突变为FUS G1566A时,相应基因矫正的gRNA-mCherry质粒为FUS-gRNA-mCherry;相应基因矫正的重组模板为序列表中序列2所示的双链DNA。所述FUS-gRNA-mCherry的构建方法如下:首先利用Phusion High-Fidelity PCR Master Mix with GC Buffer(NEB,货号为M0532)将合成的FUS gRNA oligo(FUS-gRNA-mCherry-F:5’-GAGGCCGTATTAATTAGCC-3’;FUS-gRNA-mCherry-R:5’-GGCTAATTAATACGGCCTCC-3’)退火延伸成为双链DNA,然后利用Gibson Assemly Master Mix(NEB,货号为E2611)将所述FUS gRNA双链DNA经同源重组连接进入用限制性内切酶AflII酶切纯化后的gRNA-mCherry载体(Addgene,货号为#78544),从而构建成为所述FUS-gRNA-mCherry载体。所述FUS-gRNA-mCherry的结构描述如下:将序列表中序列1所示的DNA片段(gRNA的识别区域序列,即间隔序列spacer对应的编码序列)插入到gRNA-mCherry质粒的酶切位点AflII间后得到的重组质粒。
当需要矫正的相关基因突变为SOD1 A272C时,相应基因矫正的gRNA-mCherry质粒为SOD1-gRNA-mCherry;相应基因矫正的重组模板为序列表中序列4所示的双链DNA。所述SOD1-gRNA-mCherry的构建方法如下:首先利用Phusion High-Fidelity PCR Master Mix with GC Buffer(NEB,货号为M0532)将合成的SOD1 gRNA oligo(SOD1-gRNA-mCherry-F:5’-ATGTGACTGCTGACAAAGA-3’;SOD1-gRNA-mCherry–R:5’-CTTTGGCAGCAGTCACATC-3’)退火延伸成为双链DNA,然后利用Gibson Assemly Master Mix(NEB,货号为E2611)将所述SOD1gRNA双链DNA经同源重组连接进入用限制性内切酶AflII酶切纯化后的gRNA-mCherry载体(Addgene,货号为#78544),从而构建成为所述SOD1-gRNA-mCherry载体。所述SOD1-gRNA-mCherry的结构描述如下:将序列表中序列3所示的DNA片段(gRNA的识别区域序列,即间隔序列spacer对应的编码序列)插入到gRNA-mCherry质粒的酶切位点AflII间后得到的重组质粒。
其中,所述Cas9-GFP质粒(货号:#63592)和gRNA-mCherry空载体(货号:#62340)均为Addgene公司产品。
3、电转
用步骤2中获得的电转液重悬步骤1中收集的相对应的细胞,然后转移到电转杯里,并放入电转仪(Lonza 4D nucleofector)中,进行电转,电转程序选择CM113(具体步骤参考Lonza 4D nucleofector说明书),分别得到电转后的细胞悬液(5×106细胞/100μl)。
4、分别将电转后的细胞悬液加入到matrigel包被的培养板中,用mTESR培养,放回培养箱中。电转24小时后,观察电转效率。用荧光显微镜观察绿色荧光和红色荧光,双阳性比例大于1%,即可进行下一步实验。
5、电转48小时后,TrypLE消化收集细胞,用流式细胞仪分选双阳性细胞。将分选得到的细胞置入到预先铺好经过丝裂霉素灭活处理的MEF的六孔板的孔中,用cDF12培养基培养,放回培养箱中。
6、持续用cDF12培养基培养约14天,即可看到由单细胞形成的小克隆,用CAPs方法鉴定(用PCR技术扩增出包含突变位点的片段,由于一个SNP的差异,可以用相应的限制性内切酶鉴定)。
FUS-F:5’-GAGAAAGTGGTTTCATTTTGAGGGCTAGGTGGA-3’;
FUS-R:5’-TTGTTTGAGCCTCACCATTAAAAGGGCCAAAAG-3’。
FUS鉴定用限制性内切酶:BSAXI。
当相关基因突变FUS G1566A存在时,经过FUS-F/FUS-R的PCR扩增,所得产物中含有限制性内切酶BSAXI的识别序列,因而当采用BSAXI对PCR产物进行酶切后,会产生大小约为790bp,540bp和250bp的3个条带。而当相关基因突变FUS G1566A不存在时,经过FUS-F/FUS-R的PCR扩增,所得产物中不含有限制性内切酶BSAXI的识别序列,因而当采用BSAXI对PCR产物进行酶切后,会产生大小约为540bp和250bp bp的2个条带。
SOD1-F:5’-CCCATCTTTCTTCCCAGAGCATTAGTGTGTAGACG-3’;
SOD1-R:5’-ACAAAATGTTCTGTTTAACAAGTGAGAAACCCAATCCT-3’。
SOD1鉴定用限制性内切酶:APEKI。
当相关基因突变SOD1 A272C存在时,经过SOD1-F/SOD1-R的PCR扩增,所得产物中含有限制性内切酶APEKI的识别序列,因而当采用APEKI对PCR产物进行酶切后,会产生大小约为600bp,400bp和200bp bp的3个条带。而当相关基因突变SOD1A272C不存在时,经过SOD1-F/SOD1-R的PCR扩增,所得产物中不含有限制性内切酶APEKI的识别序列,因而当采用APEKI对PCR产物进行酶切后,会产生大小约为600bp的1个条带。
经鉴定,最终成功获得将突变基因FUSG1566A矫正为FUSG1566G的克隆,突变基因SOD1A272C矫正为SOD1A272A的克隆。即成功获得两种经过基因矫正的iPSC,分别简称为FUSG1566G-iPSC和SOD1A272A-iPSC。
具体的基因矫正方案和鉴定结果如图2中A、B和C所示。
实施例3、携带ALS致病基因突变的运动神经元和经基因矫正的运动神经元的获得及鉴定
一、携带ALS致病基因突变的运动神经元和经基因矫正的运动神经元的获得
将实施例1中得到的两种携带基因突变的iPSC(FUSG1566A-iPSC和SOD1A272C-iPSC)和实施例2中得到的两种经过基因矫正的iPSC(FUSG1566G-iPSC和SOD1A272A-iPSC)同时分别定向诱导分化为运动神经元,分别得到FUSG1566A-MN,SOD1A272C-MN,FUSG1566G-MN,SOD1A272A-MN。iPSC定向分化成运动神经元细胞的方案示意图如图3中A所示。具体操作如下:
1、将实施例1和实施例2获得的四种iPSC的小克隆接种到预先铺好经过丝裂霉素灭活处理的MEF的培养板中,用cDF12培养基培养大约1-2天,至其密度达到20%左右,得到待分化的细胞。
2、将待分化的细胞用MN培养基1继续培养,每天换液,持续培养3天,得到1次培养后的细胞。
3、将1次培养后的细胞用MN培养基2继续培养,每天换液,持续培养3天,得到2次培养后的细胞。
4、将2次培养后的细胞用MN培养基3继续培养,每天换液,持续培养1天,得到3次培养后的细胞。
5、将3次培养后的细胞以单细胞转到Matrigel包被的培养板中用MN诱导培养基4进行培养,持续培养1天,得到4次培养后的细胞。在该步骤中,还可于传代前后用Rock inhibitor处理以提高细胞存活率。
6、将4次培养后的细胞用MN培养基5进行培养,持续培养3天,能观察到明显的抽丝,得到形态较好的运动神经元细胞。
FUSG1566A-MN,SOD1A272C-MN,FUSG1566G-MN,SOD1A272A-MN运动神经元细胞形态如图3中B所示。
二、携带ALS致病基因突变的运动神经元和经基因矫正的运动神经元的鉴定
利用免疫荧光检测技术检测步骤一获得的FUSG1566A-MN,SOD1A272C-MN,FUSG1566G-MN,SOD1A272A-MN的运动神经元标记物HB9和ISL1的表达情况,以及神经元细胞标记物MAP2的表达情况。具体实验步骤参照实施例1中步骤二1。
一抗及稀释比例如下:抗人HB9抗体(DSHB公司,货号81.5C10,1:50),抗人ISL1抗体(abcam,ab20670,1:200),抗人MAP2抗体(Sigma公司,货号T2220,1:500)。
检测结果如图3中C所示:可见携带基因突变的iPSC和清除基因突变的iPSC都可以定向分化为MN细胞,得到的MN细胞均表达HB9,ISL1,MAP2标记物。
实施例4、携带ALS致病基因突变的运动神经元和经基因矫正的运动神经元之间差异基因表达的转录组分析
分别收集实施例3中获得的携带ALS致病基因突变的运动神经元和经基因矫正的运动神经元,即FUSG1566A-MN/FUSG1566G-MN和SOD1A272C-MN/SOD1A272A-MN。提取RNA后进行转录组测序分析(转录组分析由北京诺禾致源科技股份有限公司完成)。在分析由于基因突变FUSG1566A导致的差异基因和基因突变SOD1A272C导致的差异基因的基础上,进一步比较二者共同的差异。
实验结果如图4中A、B、C所示:FUSG1566A-MN/FUSG1566G-MN组,下调基因13个,上调基因33个;SOD1A272C-MN/SOD1A272A-MN组,下调基因575个,上调基因364个。然后将两组进一步比较发现两组共同下调基因6个,共同上调基因1个。
- 还没有人留言评论。精彩留言会获得点赞!