利奥西呱中间体的制备方法与流程
本发明属于药物合成领域,具体涉及一种利奥西呱中间体的制备方法。
背景技术:
利奥西呱(riociguat),化学名:n-[4,6-二氨基-2-[1-[(2-氟苯基)甲基]-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基]-n-甲基氨基甲酸甲酯,是首个新型的可溶性的鸟苷酸环化酶(sgc)激动剂,可通过不依赖no的结合位点直接激活sgc从而催化cgmp的合成。本品由德国拜耳公司研发,主要用于肺动脉高压(pah)和慢性血栓栓塞性肺高压(cteph),2013年12月首次在美国批准上市,其结构式如下:
制备利奥西呱的方法已经有多种,主要集中在先制备得到中间体2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]嘧啶-4,5,6-三胺,再经过酰基化、甲基化反应得到利奥西呱成品。根据所选用的起始原料不同,主要分为以下几种路线:
cn1330649c报道以1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-甲脒盐酸盐和(苯基亚肼基)丙二腈为起始原料,经环合反应得到2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-[(e)苯基二氮烯基]-4,6-嘧啶二胺,然后经过雷尼镍高压催化氢化还原制备2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺,成盐纯化后与氯甲酸甲酯发生酰胺化反应,最后与碘甲烷发生甲基化反应得到目标产物利奥西呱,具体合成路线如下:
该方法以雷尼镍为催化剂,通过高温高压催化氢化的方法还原制备2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺,雷尼镍活性较高,在空气中易燃,不利于车间安全生产,使用高温高压催化氢化工艺对设备要求较高,长期使用容易导致设备疲劳损坏,不利于车间安全生产。2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺的后处理使用5n盐酸成盐,稀碳酸氢钠溶液游离,乙酸乙酯萃取,浪费了大量物料,降低了生产收率(质量收率低于60%)。
cn102791707a报道了不需要成盐方法制备2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺的工艺,同时使用二碳酸二甲酯替代了剧毒的氯甲酸甲酯作为酰胺化试剂,最后与碘甲烷发生甲基化反应得到利奥西呱粗品,具体合成路线如下:
该方法使用钯炭为催化剂,通过高温高压催化氢化的方法还原制备2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺。但该工艺仍然没有避免使用高温高压催化氢化设备,同时后处理需要浓缩大量的dmf溶液,对设备要求较高,不利于车间安全生产。
cn102939289报道了一条新的工艺制备2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺。该路线3-碘-1h-吡唑并[3,4-b]吡啶为起始原料,与5-氯-2-硝基苯-1,3-二胺缩合后,经过还原反应制备2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺,具体合成路线如下:
该方法使用钯炭为催化剂,通过常压催化氢化,避免使用高温高压催化氢化设备,但是所用起始原料较难得到,很难商业化购买,不适合工业化生产。同时,缩合反应所用钯和锡试剂剧毒,在医药领域是绝对慎重使用的,不适宜工业化生产。
cn105461715a报道了一种常压下催化加氢制备高纯度利奥西呱中间体2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺的合成工艺,包括首先将1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-甲脒盐酸盐、苯丙偶氮丙二腈和强碱加入到反应瓶中,然后加入dmf,升温至100~120℃反应10~15h,降至10~30℃,经后处理得到2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-(e)苯基二氮烯基]4,6-嘧啶二胺;接着将所得化合物、氯化铵和铁粉加入到反应瓶中,加入乙醇/水混合溶液,加热至回流反应6~10h,降至10~30℃,经处理后得到2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺,具体合成路线如下:
该方法使用铁粉-氯化铵为还原剂,避免使用高温高压催化氢化设备,但是铁粉在乙醇/水体系中会产生大量的铁泥,且与水反应生成粘稠的固体盐,难以过滤,后处理非常困难;同时氯化铵容易导致残渣增加,使得产品纯度降低;后处理采用高温65℃浓缩乙醇-水,导致杂质增加,不适合工业化生产。
综上所述,现有的合成利奥西呱中间体2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺的合成路线中,存在各种各样的不足,诸如制备工艺复杂、起始原料不易得、合成路线总收率低、需要使用高温高压催化氢化设备、后处理难、不适合工业化生产,因此,开发一种简便、低成本、适宜工业化生产的利奥西呱中间体的合成方法是十分必要并且具有经济实用价值的。
技术实现要素:
本发明的目的在于解决现有技术的不足,提供了一种简便合成高纯度利奥西呱中间体2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺(以下简称化合物iv)的方法。
为了达到上述发明目的,本发明采用如下技术方案:
一种利奥西呱中间体化合物iv的制备方法,包括:化合物iii在醇溶剂中经催化还原反应得到式化合物iv;
其中所述还原试剂选自水合肼或甲酸铵。
在一些实施方式中,所述水合肼为70%-85%水合肼溶液,例如85%水合肼溶液。
在一些实施方式中,所述催化试剂选自雷尼镍、pt/碳或pd/碳,例如10%的pd/碳。
在一些实施方式中,所述化合物iii与还原试剂的摩尔比为1:30~300,例如1:40~50。
在一些实施方式中,所述化合物iii与催化剂的质量比为1:0.3~1.1,例如1:0.4~0.5。
在一些实施方式中,所述醇溶剂选自c1–c5醇,例如甲醇、乙醇、丙醇、异丙醇、丁醇或戊醇。
在一些实施方式中,所述反应温度为50℃~85℃,例如70℃~80℃。
在一些实施方式中,反应结束后,后处理还包括对化合物iv进行纯化的步骤,一般采用无水乙醇纯化,例如采用无水乙醇搅拌打浆。
本发明所述化合物iv的制备方法,对反应时间无特别要求,一般依据原料的用量及反应程度,通常反应时间为10h-24h,优选10h-12h。
本发明所述化合物iv的制备方法,采用本领域的常规方法将析出的固体进行分离和干燥。所述分离,采用本领域的常规方法例如过滤、离心等;所述干燥,采用本领域的常规方法例如自然干燥、鼓风干燥或减压干燥,优选鼓风干燥;干燥温度约45℃~55℃;干燥时间为5~12小时,优选为7~8小时。
本发明还提供了化合物iii的制备方法,包括以下步骤:以1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-甲脒盐酸盐(化合物i)和(苯基亚肼基)丙二腈(化合物ii)为起始原料,在醇钠作用下,经环合反应得到2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-[(e)苯基二氮烯基]-4,6-嘧啶二胺(化合物iii),具体反应式如下:
在一些实施方式中,所述醇钠为甲醇钠溶液或乙醇钠溶液,例如30%~50%的甲醇钠溶液或50%~80%的乙醇钠溶液。
在一些实施方式中,所述化合物i、化合物ii、醇钠的摩尔比为1:0.9~1.5:0.9~1.5,例如1:0.9~1.1:0.9~1.1或1:1.0:1.0。
在一些实施方式中,所述反应溶剂选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dma)或n-甲基-2-吡咯烷酮(nmp)。
在一些实施方式中,所述反应温度95℃~135℃,例如105℃~115℃。
本发明的有益效果是:
(1)化合物iii的制备过程中,采用醇钠溶液替换固体醇钠,解决了现有技术中所得化合物iii的产品杂质多,某些杂质容易带到最终产品,导致最终产品新增杂质多,纯化困难的问题。
(2)化合物iv的制备中,采用水合肼或甲酸铵作还原剂,该反应属于常压反应,提高了工业化生产安全性,避免使用高温高压催化氢化设备,降低了工艺对设备的要求,极大简化工艺操作。
(3)使用安全性更高的醇溶剂作为溶剂,避免了使用易燃易爆和有毒试剂,减少了对操作员工的危害,有利于工业化生产。
(4)化合物iv后处理使用乙醇打浆纯化,提高了中间体iv的纯度,可直接用于合成利奥西呱的后续步骤。
(5)本发明制备得到的产品收率高、纯度高。
附图说明
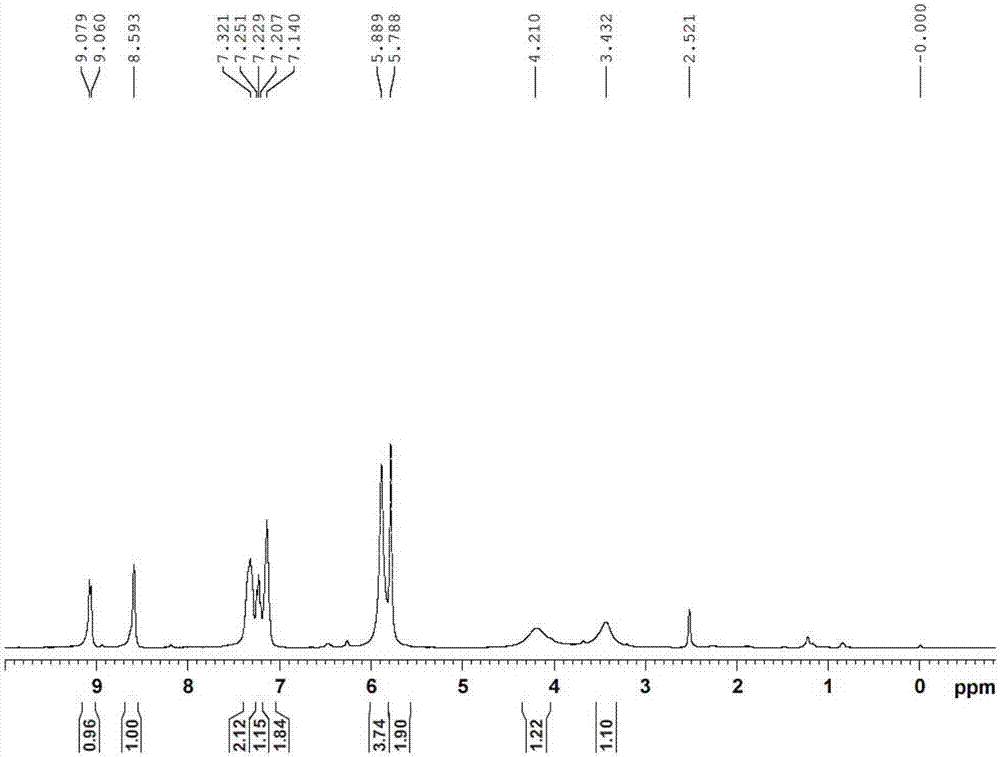
图1是化合物iii的1h核磁共振谱。
图2是化合物iv的1h核磁共振谱。
具体实施方式
为体现本发明的技术方案及其所取得的效果,下面将结合具体实施例对本发明做进一步说明,但本发明的保护范围并未局限于具体实施例。
实施例一化合物iii的制备
2l反应瓶中依次加入n,n-二甲基甲酰胺1.5l,化合物i(250g,0.82mol),化合物ii(140g,0.82mol),30%甲醇钠溶液(150ml,0.83mol),反应液升温至105~115℃,保温反应15~16小时。反应液加入10l纯化水中,室温搅拌4~5小时。过滤,滤饼置于鼓风干燥箱中,控制温度50~60℃,干燥23~24小时,得到化合物iii红褐色固体356g(0.81mol),摩尔收率98.8%,hplc纯度99.5%。
实施例二化合物iii的制备
2l反应瓶中依次加入n,n-二甲基乙酰胺1.5l,化合物i(250g,0.82mol),化合物ii(140g,0.82mol),50%乙醇钠溶液(115ml,0.83mol),反应液升温至110~115℃,保温反应14~16小时。反应液加入10l纯化水中,室温搅拌4~5小时。过滤,滤饼置于鼓风干燥箱中,控制温度50~60℃,干燥23~24小时,得到化合物iii红褐色固体351g(0.80mol),摩尔收率97.6%,hplc纯度99.2%。
实施例三化合物iv的制备
1l反应瓶中依次加入无水乙醇750ml,化合物iii(15.0g,34mmol),85%水合肼溶液(80ml,1.36mol),搅拌下加入10%钯炭7.5g,加热至70~80℃,保温70~80℃反应12小时。反应液过滤,滤液减压浓缩至干,加入无水乙醇100ml搅拌2小时。过滤,滤饼45~55℃鼓风干燥7~8小时,得化合物iv类白色固体9.95g(28.4mmol),摩尔收率83.5%,hplc纯度99.4%,重金属含量<20ppm。
实施例四化合物iv的制备
1l反应瓶中依次加入无水乙醇500ml,化合物iii(10g,22.7mmol)),85%水合肼溶液(400ml,7.0mol),加热至70~80℃,保温70~80℃反应12小时。反应液过滤,滤液减压浓缩至干,加入无水乙醇60ml搅拌打浆2小时。过滤,滤饼45~55℃鼓风干燥7~8小时,得化合物iv类白色固体6.37g(18.2mmol),摩尔收率80.2%,hplc纯度99.0%。
实施例五化合物iv的制备
1l反应瓶中依次加入无水乙醇750ml,化合物iii(15.0g,34mmol),70%水合肼溶液(95ml,1.37mol),搅拌下加入10%钯炭7.5g,加热至70~80℃,保温70~80℃反应12小时。反应液过滤,滤液减压浓缩至干,加入无水乙醇100ml搅拌2小时。过滤,滤饼45~55℃鼓风干燥7~8小时,得化合物iv类白色固体9.87g(28.2mmol),摩尔收率83.0%,hplc纯度99.4%,重金属含量<20ppm。
实施例六化合物iv的制备
1l反应瓶中依次加入无水甲醇750ml,化合物iii(15g,34mmol),85%水合肼溶液(80ml,1.36mol),搅拌下加入10%钯炭7.5g,加热至50~65℃保温反应10小时。反应液过滤,滤液减压浓缩至干,加入无水乙醇100ml搅拌打浆2小时。过滤,滤饼45~55℃鼓风干燥7~8小时,得化合物iv类白色固体9.84g(28.1mmol),摩尔收率82.8%,hplc纯度99.3%,重金属含量<20ppm。
实施例七化合物iv的制备
1l反应瓶中依次加入无水甲醇750ml,化合物iii(15g,34mmol),甲酸铵(107g,1.7mol),搅拌下加入10%钯炭7.5g,加热至50~65℃,保温反应10小时。反应液过滤,滤液减压浓缩至干,加入无水乙醇100ml搅拌打浆2小时。过滤,滤饼45~55℃鼓风干燥7~8小时,得化合物iv类白色固体9.60g(27.4mmol),摩尔收率80.6%,hplc纯度99.0%,重金属含量<30ppm。
实施例八化合物iv的制备
1l反应瓶中依次加入无水乙醇750ml,化合物iii(15g,34mmol),85%水合肼溶液(80ml,1.36mol),搅拌下加入雷尼镍6.0g,加热至70~80℃,保温70~80℃反应12小时。反应液过滤,滤液减压浓缩至干,加入无水乙醇100ml搅拌打浆2小时。过滤,滤饼45~55℃鼓风干燥7~8小时,得化合物iv类白色固体9.77g(27.9mmol),质量收率82.0%,hplc纯度为99.2%,重金属含量<20ppm。
- 还没有人留言评论。精彩留言会获得点赞!