刺槐素‑7‑O‑葡萄糖醛酸苷的制备方法与流程
本发明涉及一种从香青兰中提取分离得到刺槐素-7-O-葡萄糖醛酸苷的方法。
背景技术:
刘金旗(贡菊化学成分的研究[J].中国中药杂志,2001,26(8):547-548.)首次报道从贡菊植物分离得到刺槐素-7-O-葡萄糖醛酸苷的结构,该工艺采用萃取、硅胶柱色谱、聚酰胺柱色谱、重结晶等技术联合从2.5kg贡菊中分离得到200mg的刺槐素-7-O-葡萄糖醛酸苷,操作复杂、步骤繁琐。
刺槐素-7-O-葡萄糖醛酸苷的结构式为:
杨建波(新疆一枝蒿化学成分的研究[J].中草药,2008,39(8):1125-1127.)采用乙醇回流提取、萃取、硅胶柱、聚酰胺柱、SephadexLH-20柱、ODS柱反复柱层析分离得到刺槐素-7-O-葡萄糖醛酸苷,工艺操作步骤繁琐。
张现涛(梓木草的化学成分[J].Chinese Journal of Natural Medicines,2005,3(6):357-358.)采用乙醇回流提取、萃取、硅胶柱、SephadexLH-20柱、ODS柱反复柱层析分离得到刺槐素-7-O-葡萄糖醛酸苷,该二艺操作步骤繁琐。
靳鑫(穿心莲化学成分的研究,中草药,2012,43(1):47-50)采用萃取、硅胶柱色谱等手段首次从穿心莲植物分离得到刺槐素-7-O-葡萄糖醛酸苷,该工艺采用了6种技术联合进行分离,操作复杂、步骤繁琐。
黄松(独脚金黄酮类化学成分研究[J].中药材,2010,33(7):1089-1091.)采用D101大孔树脂、聚酰胺柱层析和反相制备液相色谱法从1kg独角金植物中分离得到9.46mg的刺槐素-7-O-葡萄糖醛酸苷,该方法分离制备得到刺槐素-7-O-葡萄糖醛酸苷的量较少,仅为9.64mg·kg-1。
目前无市售的刺槐素-7-O-葡萄糖醛酸苷纯品,其药理活性尚未见报道。
技术实现要素:
本发明的目的是提供一种从香青兰中提取分离得到刺槐素-7-O-葡萄糖醛酸苷的方法。
本发明的目的是通过以下技术方案实现的:
a、将香青兰干燥的地上部分粉碎,用5~10倍量的40%~75%乙醇浸泡12~24h;
b、采用渗漉法,香青兰药材与40%~75%乙醇比例1∶30~40,流速1~6mL/min,收集前15倍量的渗漉液;
c、渗漉液用孔径0.1~10mm的网孔过滤1~5次,滤液合并;
d、用减压浓缩装置,在压力:0.08~0.1Mpa,温度:30~60℃下,蒸馏除去乙醇得0.8~1.2g/mI的稠浸膏;水浴干燥后得浸膏粉;
e、将浸膏粉采用70%~100%乙醇溶解后干法拌样,上100~200目聚酰胺进行柱层析,20~95%乙醇梯度洗脱,并收集40%~60%乙醇的洗脱液;洗脱液用减压蒸馏装置,在压力:0.08~0.1Mpa,温度:40~60℃下蒸干,得浸膏粉;
f、将上述所得浸膏粉用50%甲醇溶解上ODS填料,50~100%甲醇-0.5%甲酸水梯度洗脱,并收集55~60%甲醇-0.5%甲酸水的洗脱液;洗脱液用减压蒸馏装置,在压力:0.08~0.1Mpa,温度:40~60℃下蒸干得化合物I粗品。
g、为了进一步纯化所得粗品,滤去杂质,采用HPLC在流动相为55~60vol%甲醇和0.5%甲酸水溶液的条件下对粗品进行制备得制备液,洗脱液用减压蒸馏装置,在压力:0.08~0.1Mpa,温度:40~60℃下蒸干得单体刺槐素-7-O-葡萄糖醛酸苷。
有益技术效果
本专利采用聚酰胺和ODS柱色谱技术联合的方法,方法简单,步骤简短。从1kg香青兰植物中分离得到121mg的刺槐素-7-O-葡萄糖醛酸苷,转移率较高。
本发明获得的刺槐素-7-O-葡萄糖醛酸苷纯度高,达到98%。
附图说明
下面将结合附图和具体实施方式对本发明的上述和其他目的、特征和优点作出进一步详细的说明。
图1为本发明提供方法的工艺流程图;
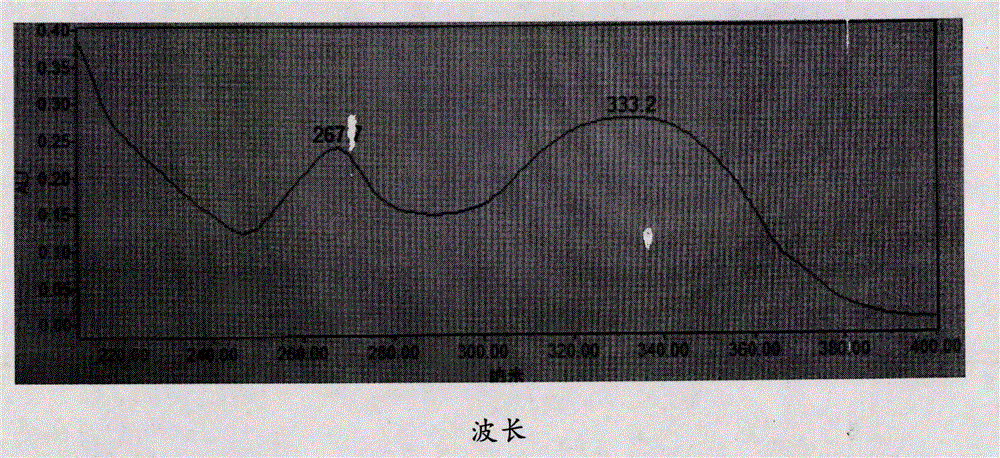
图2为根据本发明一个实施例所制备的刺槐素-7-O-葡萄糖醛酸苷的紫外光谱(UV)图;
图3为根据本发明一个实施例所制备的刺槐素-7-O-葡萄糖醛酸苷的红外光谱(IR)图;
图4为根据本发明一个实施例所制备的刺槐素-7-O-葡萄糖醛酸苷的电喷雾质谱(ESI-MS)图;
图5为根据本发明一个实施例所制备的刺槐素-7-O-葡萄糖醛酸苷的核磁共振氢谱(1H-NMR)图;
图6为根据本发明一个实施例所制备的刺槐素-7-O-葡萄糖醛酸苷的核磁共振碳谱(13C-NMR)图。
具体实施方式
本发明提供的一种刺槐素-7-O-葡萄糖醛酸苷的制备方法,包括获得香青兰的提取物,以及对提取物进一步层析、HPLC制备等步骤。下面结合附图,详述本发明所提供的方法。
如图1所示,首先,将1份重量的香青兰干燥的地上部分粉碎,用30~40重量倍数的40~75vol%的乙醇溶液渗漉提取。然后,将提取液浓缩,例如可以采用减压浓缩的方法,浓缩后提取液呈膏状。之后再将膏状物干燥,例如可以采用真空干燥的方法,将膏状物制成浸膏粉。
将浸膏粉进行聚酰胺柱层析。层析的步骤可以为:将浸膏粉采用高浓度醇溶解后拌入聚酰胺中,例如可以采用100~200目的层析用聚酰胺,拌入的比例为浓缩后的浸膏粉与层析用聚酰胺的重量比为1∶10~20。上柱完毕后,流动相采用乙醇溶液,按梯度20%~95%的比例通入流动相进行洗脱。合并相同组分洗脱液,浓缩,得到40%~60%乙醇洗脱馏分。
将40%~60%乙醇洗脱馏分采用70%乙醇溶解,滤除杂质,再浓缩后进行ODS柱层析,流动相采用复合洗脱剂,所述复合洗脱剂由A组分和B组分构成,所述A组分选自下述组中的至少一种:甲醇、乙醇、丙酮、异丙酮或丁醇,所述B组分为水、甲酸-水、磷酸-水,所述A组分与B组分的体积比为1∶1~8∶2。例如流动相可以采用甲醇-0.5%甲酸水,甲醇-0.5%甲酸水的体积比依次为55∶45、6∶4、8∶2。此时刺槐素-7-O-葡萄糖醛酸苷的组分主要在流动相甲醇-0.5%甲酸水体积比为6∶4时被洗脱。最后,合并相同组分洗脱液并浓缩,得到化合物I的粗提物。
进一步地,为了提高刺槐素-7-O-葡萄糖醛酸苷的纯度,还可用HPLC对刺槐素-7-O-葡萄糖醛酸苷进行制备,例如色谱柱为C18(150*10mm,10μm),流动相为50~60vol%甲醇和0.5%甲酸水溶液,流速为3.5mL/min,进样量为0.5~1.0mL,对上述粗提物进行HPLC制备,即得到淡黄色粉末状结晶,为刺槐素-7-O-葡萄糖醛酸苷成品,其纯度可达95%以上。经UV、IR、MS、1H-NMR、13C-NMR对所得组分的结构进行鉴定,可知其为刺槐素-7-O-葡萄糖醛酸苷。
图2、图3为根据本发明的制备方法制备的刺槐素-7-O-葡萄糖醛酸苷的UV图谱和IR图谱。
图4为根据本发明的制备方法制备的刺槐素-7-O-葡萄糖醛酸苷的ESI-MS的分析可知,ESI-MS特征峰在m/z461[M+](正离子),m/z316.20[M+-COOH],m/z285.10[M+-Glucuronide]。再结合图5、图6所示的根据本发明的制备方法制备的田蓟苷的NMR图谱分析,可确定其分子量为460,分子式为C22H20O11,其中,1H-NMR(400MHz,DMSO)图谱中表现出典型的黄酮苷类化合物的氢谱特征,通过耦合常数及化学位移可确定每个氢的归属,δ:12.91(5-OH),8.07(d,J=8.0Hz,H-2′,6′),7.13(d,J=8.4Hz,H-3′,5′),6.95(s,H-3),6.86(d,J=1.5Hz,H-8),6.45(d,J=1.5Hz,H-6),5.12(d,J=7.5Hz,H-1″);13C-NMR(DMSO,400MHz)图谱中呈现出22个碳信号,为1个CH3,12个CH,8个季碳,δc表明分子中含一个羰基(δ182.9),δ:163.83(C-2),103.79(C-3),182.06(C-4),163.10(C-7),162.47(C-4′),157.76(C-9),128.47(C-2′,6′),122.68(C-1′),114.66(C-3′,5′),105.37(C-10),99.93,99.57(C-6),(C-1″),94.81(C-8),73.28(C-2″),76.40(C-3″),72.30(C-4″),74.05(C-5″),172.20(C-6″),55.60(OCH3)。通过归峰解谱可确定所得化合物为刺槐素-7-O-葡萄糖醛酸苷。
下面通过具体实施例对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围。
实施例1:从香青兰中提取分离得到刺槐素-7-O-葡萄糖醛酸苷的方法,按以下步骤进行:
a、将香青兰干燥的地上部分粉碎,用5倍量的40%的乙醇浸泡12h,得到提取液;
b、采用渗漉法,香青兰药材3kg与40%乙醇比例1∶30,流速1mL/min,收集前15倍量的乙醇渗漉液;
c、渗漉液用孔径0.1mm的网孔过滤1次,滤液合并;
d、用减压浓缩装置,在压力:0.08Mpa,温度:30℃下,蒸馏除去乙醇得0.8g/mL的稠浸膏;水浴干燥后得浸膏粉250g;
e、将100g浸膏粉采用70%乙醇溶解后干法拌样,上100~200目聚酰胺进行柱层析,20~95%乙醇梯度洗脱得到洗脱液共95000mL,收集50%-60%乙醇梯度的洗脱液25000mL;洗脱液用减压蒸馏装置,在压力:0.08Mpa,温度:40℃下蒸干,得浸膏粉11.00g;
f、将上述所得浸膏粉1.50g用50%甲醇溶解上ODS填料,粒径大小:50μm,孔径大小:120A,55~100%甲醇-0.5%甲酸水2L梯度洗脱得到洗脱液,收集55-60%甲醇-0.5%甲酸水梯度洗脱液800mL;洗脱液用减压蒸馏装置,在压力:0.08Mpa,温度:40℃下蒸干得化合物I的粗品;
g、为了进一步纯化所得粗品,滤去杂质,采用HPLC在流动相为55~60vol%甲醇和0.5%甲酸水溶液的条件下对粗品进行制备得制备液。制备液用减压蒸馏装置,在压力:0.08Mpa,温度:40℃下蒸干得淡黄色粉末20mg。
一、结构鉴定:取所得样品分别对UV、IR、MS、NMR进行归峰解谱,确定实施例1所提纯的样品为刺槐素-7-O-葡萄糖醛酸苷。
二、纯度鉴定:
熔点(mp)测定:255℃~257℃。
薄层色谱检测:采用两种展开体系进行检测,薄层板用聚酰胺薄膜。展开剂A为乙醇-甲酸水(体积比为1∶1),RfA为0.21;展开剂B为乙酸乙酯-95%乙醇(体积比为6∶4),RfB为0.38,紫外365nm下见黄色斑点,无杂质点。
HPLC检测:用HPLC外标法测定含量,选定260nm和330nm为检测波长。选用乙腈-0.1%磷酸水溶液(30∶70,V/V)为流动相,以刺槐素-7-O-葡萄糖醛酸苷。精密称取实施例1制备的淡黄色粉末2.50mg于250mL容量瓶,用甲醇溶解并定容至刻度线。经Waters分析型高效液相色谱检测,外标法定量,其纯度为98.36%。
得率计算:
刺槐素-7-O-葡萄糖醛酸苷浓度为9.83μg·mL-1,香青兰中刺槐素-7-O-葡萄糖醛酸苷的量为287mg/kg。
得率=(9.83μg·mL-1×250mL)/(2.5mg×3000g/367mg×287mg/kg)×100%=41.90%
经检测,实施例1提取得到的刺槐素-7-O-葡萄糖醛酸苷的纯度为98.36%,得率为41.90%。
实施例2:从香青兰中提取分离得到刺槐素-7-O-葡萄糖醛酸苷的方法,按以下步骤进行:
a、将香青兰干燥的地上部分粉碎,用10倍量的75%的乙醇浸泡24h;
b、采用渗漉法,香青兰药材4kg与75%乙醇比例1∶40,流速6mL/min,收集前15倍量乙醇渗漉液;
c、渗漉液用孔径10mm的网孔过滤5次,滤液合并;
d、用减压浓缩装置,在压力:0.1Mpa,温度:60℃下,蒸馏除去乙醇得1.2g/mL的稠浸膏;水浴干燥后得浸膏粉305g;
e、将100g浸膏粉采用70%乙醇溶解后干法拌样,上100~200目聚酰胺进行柱层析,30~75%乙醇80000mL梯度洗脱得到洗脱液,并收集40~60%乙醇的洗脱液32000mL;洗脱液用减压蒸馏装置,在压力:0.1Mpa,温度:60℃下蒸干,得浸膏粉13.65g;
f、将上述所得浸膏粉2g用50%甲醇溶解上ODS填料,粒径大小:50μm,孔径大小:120A,50~80%甲醇-0.5%甲酸水2500mL梯度洗脱,收集55-60%甲醇-0.5%甲酸水的洗脱液1500mL;洗脱液用减压蒸馏装置,在压力:0.1Mpa,温度:40℃下蒸干得化合物I的粗品;
g、为了进一步纯化所得粗品,滤去杂质,采用HPLC在流动相为55~60vol%甲醇和0.5%甲酸水溶液的条件下对粗品进行制备得制备液。制备液用减压蒸馏装置,在压力:0.08Mpa,温度:40℃下蒸干得淡黄色粉末23.50mg。
一、结构鉴定:取所得样品分别对UV、IR、MS、NMR进行归峰解谱,确定实施例1所提纯的样品为刺槐素-7-O-葡萄糖醛酸苷。
二、纯度鉴定:
熔点(mp)测定:255℃~257℃。
薄层色谱检测:采用两种展开体系进行检测,薄层板用聚酰胺薄膜。展开剂A为乙醇-甲酸水(体积比为1∶1),RfA为0.21;展开剂B为乙酸乙酯-95%乙醇(体积比为6∶4),RfB为0.38,紫外365nm下见黄色斑点,无杂质点。
HPLC检测:用HPLC外标法测定含量,选定260nm和330nm为检测波长。选用乙腈-0.1%磷酸水溶液(30∶70,V/V)为流动相,以刺槐素-7-O-葡萄糖醛酸苷。将实施例2制备的化合物C结晶用甲醇溶解,经Waters分析型高效液相色谱检测,外标法定量,其纯度为99.15%,得率为42.09%。
实施例3:从香青兰中提取分离得到刺槐素-7-O-葡萄糖醛酸苷的方法,按以下步骤进行:
a、将香青兰干燥的地上部分粉碎,用7倍量的60%的乙醇浸泡18h;
b、采用渗漉法,香青兰药材2kg与60%乙醇比例1∶35,流速4mL/min,收集前15倍量乙醇渗漉液;
c、渗漉液用孔径0.5mm的网孔过滤2次,滤液合并;
d、用减压浓缩装置,在压力:0.09Mpa,温度:50℃下,蒸馏除去乙醇得1.0g/mL的稠浸膏;水浴干燥后得浸膏粉170g;
e、将浸膏粉100g采用80%乙醇溶解后干法拌样,上100~200目聚酰胺进行柱层析,30~95%乙醇80000mL梯度洗脱得到洗脱液,并收集50%-60%乙醇的洗脱液21000mL;洗脱液用减压蒸馏装置,在压力:0.1Mpa,温度:60℃下蒸干,得浸膏粉10.50g;
f、将上述所得浸膏粉1.50g用50%甲醇溶解上ODS填料,粒径大小:50μm,孔径大小:120A,55~80%甲醇-0.5%甲酸水2000mL洗脱,并收集55~80%甲醇-0.5%甲酸水的洗脱液1000mL;洗脱液用减压蒸馏装置,在压力:0.08Mpa,温度:40℃下蒸干得化合物I的粗品;
g、为了进一步纯化所得粗品,滤去杂质,采用HPLC在流动相为55~60vol%甲醇和0.5%甲酸水溶液的条件下对粗品进行制备得制备液。制备液用减压蒸馏装置,在压力:0.08Mpa,温度:50℃下蒸干得黄色粉末20.50mg。
一、结构鉴定:取所得样品分别对UV、IR、MS、NMR进行归峰解谱,确定实施例1所提纯的样品为刺槐素-7-O-葡萄糖醛酸苷。
二、纯度鉴定:
熔点(mp)测定:255℃~257℃。
薄层色谱检测:采用两种展开体系进行检测,薄层板用聚酰胺薄膜。展开剂A为乙醇-甲酸水(体积比为1∶1),RfA为0.21;展开剂B为乙酸乙酯-95%乙醇(体积比为6∶4),RfB为0.38,紫外365nm下见黄色斑点,无杂质点。
HPLC检测:用HPLC外标法测定含量,选定260nm和330nm为检测波长。选用乙腈-0.1%磷酸水溶液(30∶70,V/V)为流动相,以刺槐素-7-O-葡萄糖醛酸苷。将实施例3制备的化合物C结晶用甲醇溶解,经Waters分析型高效液相色谱检测,外标法定量,其纯度为98.24%,得率为41.75%。
- 还没有人留言评论。精彩留言会获得点赞!