一种PDE4抑制剂氯比普兰的制备方法与流程
本发明涉及一种PDE4抑制剂的制备方法,具体为化合物氯比普兰的制备方法。
背景技术:
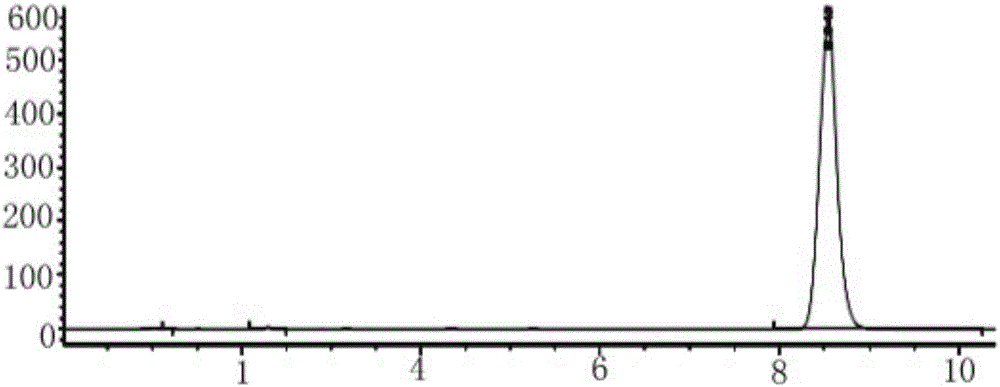
:磷酸二酯酶4(PDE4)与中枢神经系统和免疫系统的功能有密切的关系,其抑制剂被认为是作用于细胞内靶点的新抗炎药和中枢神经系统候选药物。第一代PDE4抑制剂如咯利普兰具有很好的抗抑郁效果,导致其不能上市的原因是抑制PDE4所带来的恶心呕吐等副反应。与第一代PDE4抑制剂相比,第二代PDE4抑制剂具有更好的治疗效果,但至今还没有能避免呕吐的PDE4抑制剂上市。开发出能够避免恶心呕吐反应的新型PDE4抑制剂在抑郁症和认知障碍的治疗上具有很好的应用前景。CN201210037980.3号中国专利公开了一系列的PDE4抑制剂,并证明氯比普兰(chlorbipram)在对东莨菪碱诱导的学习记忆获得性障碍大鼠模型具有很好的改善作用;对强迫游泳和悬尾行为绝望模型小鼠也具有良好的抗抑郁活性,并且已采用比格犬证明其仅有微弱甚至无致呕吐恶心副作用。该专利也同时公开了氯比普兰(chlorbipram)的合成方法,该方法采用甲基溴化后与NH类化合物对接合成制得,反应路线见Scheme1。由于NBS溴化的副反应多,溴化物不稳定,对接反应收率较低,其合成有一定的局限性,不适合该化合物的后续研发。Scheme1技术实现要素:本发明公开了一种PDE4抑制剂氯比普兰的制备方法,实验证明,该方法的产率高达71%。本制备方法的合成路线为:包括以下步骤:(1)以3-溴-4-甲氧基苯甲醛为起始原料A,还原反应生成化合物B,即3-溴-4-甲氧基苯甲醇;(2)将化合物B与间氯苯硼酸,在钯盐催化剂的催化下,进行Suzuki反应,生成化合物C;(3)将化合物C用氯化试剂进行氯化,生成化合物D;(4)将原料化合物A1与3,6-二氯哒嗪,加热至回流反应,生成化合物A2;化合物A2与醋酸钠加热回流反应,生成化合物A3;(5)将化合物D和化合物A3在碱性条件下回流反应,生成目标产物E氯比普兰。步骤(1)所述的还原反应采用的还原剂优选NaBH4。步骤(2)所述的钯盐催化剂优选四三苯基膦钯或醋酸钯或醋酸钯和配体尿素。四三苯基膦钯与化合物B的投料比优选1:20,催化剂醋酸钯与化合物B的投料比优选1:30。步骤(3)所述的氯化试剂优选四氯化碳/三苯基膦或三聚氯氰/二甲基甲酰胺体系。步骤(5)所述的回流反应溶剂优选乙二醇二甲醚或二甲基甲酰胺。本发明的制备方法相对于现有技术,具有以下优点:产率高达64-71%,较之前的合成方法产率提高42.2%-57.8%,且原料易得价廉,反应条件温和,对设备要求低,可实现产业化生产。本发明为氯比普兰的合成方法提供了一条高效易行的新途径。说明书附图图1是本实施例制备的目标化合物氯比普兰的HPLC结果图。具体实施方式下面结合实施例对本发明的制备方法作进一步的说明。但本发明的保护的范围不以以下实施例为限。实施例1(1)取30g原料A,3-溴-4-甲氧基苯甲醛,分批次缓慢加入NaBH48-10g,在乙醇中反应。通过TLC监控反应。反应完全后,除去乙醇,萃取,合并有机相,无水硫酸钠干燥,旋干得白色固体3-溴-4-甲氧基苯甲醇30.0g,产品不需要纯化即可进行下步反应,收率>98%。(2)取10g3-溴-4-甲氧基苯甲醇,加入间氯苯硼酸8.6g,碳酸钾18.8g,四三苯基膦钯1.0g,异丙醇/水溶剂100mL,室温搅拌。待固体全部溶解,除去反应体系中的氧气,无氧反应,加热,反应完后,减压旋掉异丙醇和部分水,然后用乙酸乙酯萃取,合并有机层,用无水硫酸钠干燥后用抽滤挡掉钯盐,旋干,得微黄黏稠液体化合物C11.46g,产品不需要纯化即可进行下步反应,收率>95%。(3)取化合物C11.5g(按纯品折算),加四氯化碳7.4g,分批加入三苯基膦11.0g,在二氯甲烷中,常温搅拌,TLC检测反应。反应完后,浓缩反应液,柱分离除杂,得无色透明油状液体化合物D,11.1g,收率90%。(4)加31.2g化合物A1与28.2g3.6-二氯哒嗪在乙醇中,加热至回流,TLC监控反应,反应结束后,减压旋掉乙醇得粗品,重结晶,得黄色固体化合物A244g,收率84.3%。取50g上述得到的化合物A2用醋酸/醋酸钠,搅拌加热回流,TLC检测。反应结束后除去醋酸钠,减压除去乙酸,用乙醇重结晶的淡黄色固体化合物A3,收率92%。(5)取11.0gA3于100mL圆底烧瓶中,加入11.0g化合物D,14.5g无水碳酸钾,溶剂乙二醇二甲醚,回流。反应完后,用乙酸乙酯/石油醚为洗脱剂,柱分离,得白色固体化合物E,即氯比普兰15g,收率75%。经上述方法,计算得总收率为67.5%。实施实例2:在实施例1的基础上,其他步骤不变,实施例1中步骤(2)方法变为以下方案:取10g化合物B,3-溴-4-甲氧基苯甲醇,加入间氯苯硼酸8.6g,碳酸钾18.8g,尿素82.8mg,醋酸钯210mg,异丙醇/水溶剂100mL,室温搅拌。待固体全部溶解,除去反应体系中的氧气,无氧反应,加热,反应完后,减压旋掉异丙醇和部分水,然后用乙酸乙酯萃取,合并有机层,用无水硫酸钠干燥后用抽滤挡掉钯盐,旋干,得微黄黏稠液体化合物C,11.46g,收率>95%。本实施例的总收率为65.5%。实施实例3:在实施例1的基础上,其他步骤不变,实施例1中步骤(3)方法变为以下方案:8.13g三聚氯氰,缓慢搅拌加入DMF,室温搅拌半小时左右,加入干燥的二氯甲烷,搅拌均匀,加入原料C11.5g,室温搅拌4-8小时。反应完后有机层用饱和NaCl洗涤,干燥有机层,减压旋干。粗品用柱分离的方法精置,得无色的油状氯化产物,即化合物D,11.6g(收率95%)。经计算总收率为71%。实施例4:在实施例1的基础上,其他步骤不变,实施例1中步骤(5)方法变为以下方案:取11.0gA3于100mL圆底烧瓶中,加入11.0g化合物D,14.5g无水碳酸钾,溶剂二甲基甲酰胺(DMF),回流。反应完后,用乙酸乙酯/石油醚为洗脱剂,柱分离,得白色固体化合物E14.4g,收率72%。经计算总收率为64.8%。经实验证明:步骤(2)所述的钯盐催化剂与化合物B的投料比为1:10-100的范围内,都可获得较好的收率,收率在64%以上。由上述实施例方法所得的目标化合物E,数据表征为:ESI-MS:m/z492.6([M+H]);1HNMR(400MHz,CDCl3)δ1.26(s,1H),3.62(t,J=4.8Hz,2H),3.78(s,3H),3.84(s,3H),4.14(t,J=4.8Hz,2H),5.15(s,2H),6.73(d,J=8.0Hz,1H),6.85-6.96(m,6H),7.36-7.38(m,2H),7.40(d,J=8.4Hz,1H),7.49(s,1H).13CNMR(100MHz,CDCl3)δ41.7,54.3,55.7,55.8,67.6,111.3,112.0,111.8,121.0,122.1,126.7,127.0,127.7,128.9,129.2,129.5,130.0,130.8,131.5,133.7,140.0,147.6,147.9,149.7,156.1,and157.9.HPLC结果如图1及表1所示。表1峰(#)保留时间(min)峰宽(min)峰面积(mAU·s)峰面积(%)11.1610.05115.741170.072322.2830.069216.698510.210238.5420.21597922.4497199.7176总量7944.88939本发明的制备方法与CN201210037980.3号中国专利公开的合成方法的目标产品收率比较见表2。明显可见,利用本发明方法,以3-溴-4-甲氧基苯甲醛为起始原料经四步反应,最终氯比普兰的总收率可以达到71%,一般收率也可达64-71%。而CN201210037980.3号中国专利公开的合成方法,收率只有45%。表2本专利保护的合成方法,所显示的实施实例具有一定的代表性,但是并不仅仅局限在所述的实例方法。尤其是其中Pd催化的偶联反应,反应条件不局限在Pd(PPh3)4,Pd(OAc)2及加入不同配体,不同碱及溶剂的反应条件;采用苄基醇转化为苄基氯的方法也不局限在上述实例的CCl4/PPh3和三聚氯氰/DMF两种反应方式。凡与本发明构思相同的合成方法,都应落入本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1