一种提高胞内氧化型辅酶I含量的方法与流程
本发明涉及一种提高胞内氧化型辅酶I含量的方法,属于辅因子工程技术领域。
背景技术:
辅酶I,又称烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide),是一种传递电子(氢离子)的辅酶,KEGG数据库显示NADH/NAD+作为重要的辅因子全程参与微生物细胞内的740多个代谢反应,包括氧化还原反应、能量代谢过程、调控基因表达、维持Ca+稳态以及调节细胞衰老等,尤其在糖酵解、糖异生、三羧酸循环和呼吸链中发挥着不可替代的作用。NAD+能为代谢网络中各种反应提供能量和氧化还原力,其浓度可以调控代谢网络途径,增大代谢流通量,提高氧化还原反应中生物催化效率和目标产物的产率等。
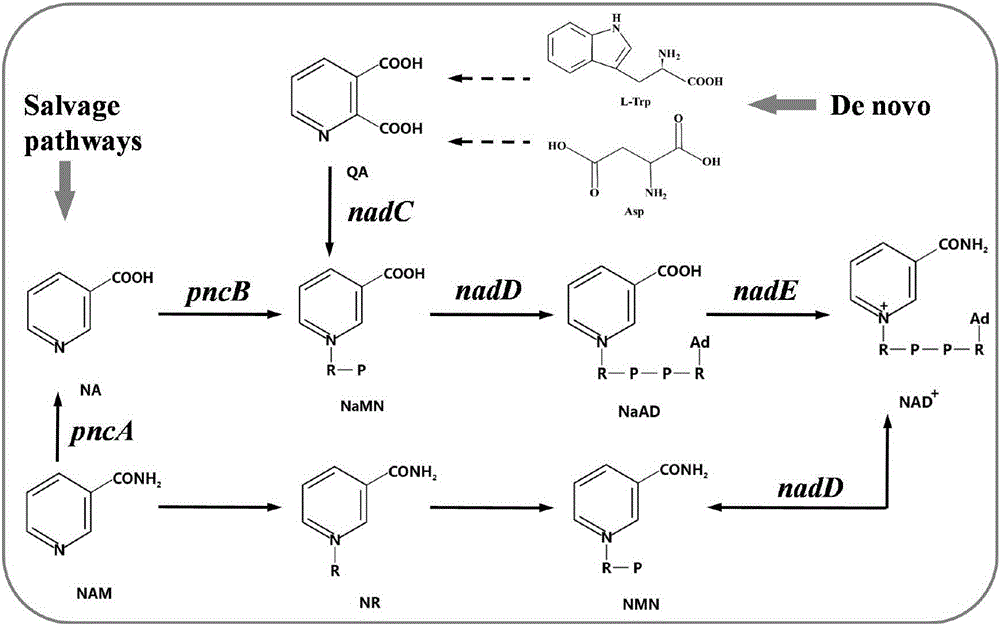
NAD+生物合成主要有两条途径:从头合成途径和补救合成途径(图1)。从头合成途径以天冬氨酸或色氨酸为前体,通过一系列步骤生成喹啉酸(QA),接着经基因nadC编码的喹啉酸磷酸核糖转移酶催化生成烟酸单核苷酸(NaMN),再由基因nadD编码的腺苷转移酶催化腺苷化生成烟酸腺嘌呤二核苷酸(NaAD),最后在基因nadE编码的NAD+合成酶催化下氨基化成NAD+。补救途径是基因pncA编码的烟酰胺酶水解烟酰胺生成烟酸,再经基因pncB编码的烟酸磷酸核糖转移酶催化烟酸生成烟酸单核苷酸(NaMN),最后生成NAD+。当细胞内大量存在NAD+前体烟酰胺(NAM)、烟酸(NA)时,补救途径对胞内NAD+的含量起到重要的作用。
目前,提高细胞内NAD+含量主要从代谢工程和生化工程两个方向上考虑,其中包括:过量表达NADH代谢相关酶、缺失NADH竞争途径、引入NADH外源代谢途径、添加外源电子受体、不同氧化还原态底物及NAD合成前体物、调节培养环境和氧化还原电势等。
大肠杆菌由于遗传背景清楚、易操作、易调控、培养基要求简单且生长周期短等优点,被广泛应用于科研工作中。然而,其细胞内的辅酶含量较低,无法起到增强相关催化反应效率的作用。因此,通过基因工程的手段增强细胞内NAD+含量对于提高胞内氧化型辅酶I的含量具有重要作用。
技术实现要素:
本发明的第一个目的是提供一种共表达pncA、pncB、nadC、nadD、nadE中至少两个基因的重组大肠杆菌。
在本发明的一种实施方式中,所述重组大肠杆菌以E.coli BL21为宿主。
在本发明的一种实施方式中,所述重组大肠杆菌是共表达pncA、pncB、nadC、nadD、nadE中的任意三种基因。
在本发明的一种实施方式中,所述重组大肠杆菌共表达pncA、nadD、nadE基因。
在本发明的一种实施方式中,所述重组大肠杆菌共表达pncB、nadE、nadD基因。
在本发明的一种实施方式中,所述重组大肠杆菌共表达nadC、nadD、nadE基因。
在本发明的一种实施方式中,所述重组大肠杆菌共表达pncB、nadE、pncA基因。
本发明的第二个目的是提供所述重组大肠杆菌的构建方法,是以pET-21a为载体,在大肠杆菌E.coli BL21中表达pncA、pncB、nadC、nadD、nadE中的至少2个基因。
本发明的第三个目的是提供一种提高重组大肠杆菌胞内氧化型辅酶I含量的方法,所述方法是将所述重组大肠杆菌接种至发酵培养基中,30~37℃培养8~72h。
在本发明的一种实施方式中,所述接种是按体积比1%的接种量进行接种。
在本发明的一种实施方式中,所述发酵培养基中还含有终浓度1~100mg/L的色氨酸、天冬氨酸、喹啉酸、烟酸、烟酰胺中的至少一种。
在本发明的一种实施方式中,所述方法是在接种后菌体OD600达到0.6-2.0时,加入终浓度为0.1-10mM的IPTG,16-37℃诱导8-48h。
本发明还提供所述重组大肠杆菌在食品、医药领域生产氧化型辅酶I方面的应用。
本发明还提供所述方法在生产含氧化型辅酶I的产品中的应用。
有益效果:本发明通过采用共表达多基因的组合,并且在发酵培养基中添加不同的前体物质,将多基因与多种类的前体物质结合,扩大代谢网络通量,大肠杆菌胞内的辅酶含量在经过代谢工程和生化工程两个手段的调控后可达到20-30μmol/g DCW。相比出发菌株BL21/pET-21a提高了近10倍,NAD+生成的效率明显提高,大肠杆菌胞内NAD+的含量也进一步增加。
附图说明
图1为辅酶NAD+合成途径示意图,其中NAD+化学结构及其相关中间体(R:核糖体、P:磷酸、Ad:腺嘌呤)化合物的缩写为:L-Trp:L-色氨酸;Asp:天冬氨酸;QA:喹啉酸;NA:烟酸;NaMN:烟酸单核苷酸;NaAD:烟酸腺嘌呤二核苷酸;NAD+:烟酰胺腺嘌呤二核苷酸;NAM:烟酰胺;NR:烟酰胺核苷;NMN:烟酰胺单核苷酸。
具体实施方式
发酵培养基:NaCl 10g/L蛋白胨10g/L,酵母提取物5g/L,pH7.2,以去离子水配制,使用前加入氨苄青霉素100μg/mL。
发酵条件:初始温度为37℃,摇床转速为200r/min,菌体OD600达到0.6-2.0时,加入终浓度为0.1-10mM的IPTG,16-37℃诱导8-48h,添加前体物质的浓度为1-100mg/L。
实施例1
(1)构建过量表达基因pncA、pncB、nadC、nadD、nadE的表达质粒:
根据NCBI上报道的大肠杆菌中基因pncA的序列(Gene ID:946276),基因pncB的序列(Gene ID:8182321),基因nadC的序列(Gene ID:948869),基因nadD的序列(Gene ID:8180157),基因nadE的序列(Gene ID:8179982)设计上下游引物,并合成带有BamH I和Xho I酶切位点的引物:
pncA-F:GGATCCATGCCCCCTCGCGCCCTGTT
pncA-R:CCGCTCGAGTTACCCCTGTGTCTCTTCCCAGTCTG
pncB-F:CGCGGATCCATGACACAATTCGCTTCT
pncB-R:CCGCTCGAGTTAACTGGCTTTTTTAATATGCGG
nadC-F:GGATCCATGCCGCCTCGCCGCTATAACC
nadC-R:CTCGAGTTAGCGAAAACGCATTGAAAGGTCGAGTG
nadD-F:CGCGGATCCATGAAATCTTTACAGGCTC
nadD-R:CCGCTCGAGTCAGCGATACAAGCCTTGTT
nadE-F:CGCGGATCCATGACATTGCAACAACAAAT
nadE-R:CCGCTCGAGTTACTTTTTCCAGAAATCATCG
提取E.coli BL21(DE3)的基因组,以其为模板,PCR克隆扩增得到目的基因pncA、pncB、nadC、nadD和nadE片段,反应条件为:98℃预变性30s;98℃变性10s;55℃退火30s;72℃分别延伸42s、80s、58s、42s、55s,30个循环;72℃延伸10min。
将目的基因产物纯化后,连接到pMD-19T载体,转化筛选阳性克隆并测序。再将载体pET-21a和T载上的目的基因用限制性核酸内切酶BamH I和Xho I酶切90min,经琼脂糖凝胶电泳回收纯化目的基因片段和质粒骨架,在T4连接酶作用下16℃连接过夜。然后将连接产物转化JM109克隆宿主感受态细胞,在氨苄青霉素抗性平板上筛选阳性克隆转化子。
(2)将质粒pET-21a-pncA、pET-21a-pncB、pET-21a-nadC、pET-21a-nadD、pET-21a-nadE导入E.coli BL21(DE3)感受态,得到重组菌株。
(3)对重组菌株进行发酵培养,接种量为按体积的1%,IPTG诱导浓度为0.1-10mM,测定其胞内的NAD+含量(表1)。其中效果比较明显的有:E.coli BL21/pET-21a-pncB和E.coli BL21/pET-21a-nadE。其胞内的辅酶含量分别达到12.475μmol/g DCW和13.398μmol/g DCW,与对照相比提高了3倍和3.3倍。
表1过表达单个基因重组菌株生长情况及NAD+含量
实施例2
(1)构建共表达多基因的大肠杆菌重组菌株:
根据已经构建成功的质粒pET-21a-pncA、pET-21a-pncB、pET-21a-nadC、pET-21a-nadD、pET-21a-nadE设计上下游引物,并且在两个多个基因之间加上SD-AS序列(AGAAGGAGATATACA),采用重叠延伸PCR技术,实现多个基因在同一个质粒上的串联表达。
合成带有BamH I和Xho I酶切位点的引物,以质粒pET-21a-pncA、pET-21a-pncB、pET-21a-nadC、pET-21a-nadD、pET-21a-nadE为模板,重叠延伸PCR克隆扩增得到目的基因pncA-pncB、pncA-nadD、pncA-nadE、pncB-nadD、pncB-nadE、nadD-nadE、nadC-nadD、nadC-nadE以及pncA-pncB-nadD、pncA-nadD-nadE、pncB-nadD-nadE、nadC-nadD-nadE、pncA-pncB-nadE片段,反应条件为:98℃预变性30s;98℃变性10s;60℃退火15s;72℃分别延伸112s、80s、90s、112s、125s、90s、95s、105s,以及155s、130s、165s、145s、165s,15个循环;72℃延伸10min,再加入上下游引物各1μL,继续上述条件,15个循环,72℃延伸10min,12℃30min。
将目的基因产物纯化后,连接到pMD-19T载体,转化筛选阳性克隆并测序。再将载体pET-21a和T载上的目的基因用限制性核酸内切酶BamH I和Xho I酶切90min,经琼脂糖凝胶电泳回收纯化目的基因片段和质粒骨架,在T4连接酶作用下16℃连接过夜。然后将连接产物转化JM109克隆宿主感受态细胞,在氨苄青霉素抗性平板上筛选阳性克隆转化子。
(2)将以上所得到的质粒导入E.coli BL21(DE3)感受态,得到多基因共表达的重组菌株
(3)对多基因共表达重组菌株进行发酵培养,先用LB氨苄青霉素平板活化重组菌株,挑取单菌落接入5mL含有终浓度100μg/mL氨苄青霉素的LB试管中,培养12h后再按1%的接种量转接于50mL的LB发酵培养基中,37℃、200r/min培养至OD600为0.6-2.0时添加终浓度为0.1-10mmol/L的IPTG,16-37℃,200r/min诱导,蛋白表达正常后进行胞内NAD+含量的测定(表2)。
表2多基因共表达重组菌株胞内NAD+含量
多基因共表达重组菌株中,E.coli BL21/pET-21a-nadE-pncB、E.coli BL21/pET-21a-pncB-nadE-nadD和E.coli BL21/pET-21a-pncB-nadE-pncA胞内NAD+含量能达到18μmol/g DCW。相比出发菌株E.coli BL21/pET-21a提高了近5倍。
实施例3
(1)配制功能因子促进剂:将色氨酸、天冬氨酸、喹啉酸、烟酸、烟酰胺分别用灭菌后的超纯水溶解,配制为终浓度100mg/L的溶液。
(2)对初始菌株E.coli BL21/pET-21a进行发酵培养,采用50ml摇瓶有氧发酵。按1%接种量从冻存管里接入5mL的LB试管中,同时接入终浓度为100μg/mL的氨苄青霉素,培养12h后再按1%的接种量转接于50mL的LB发酵培养基中,分别添加终浓度为40mg/L的功能因子促进剂,37℃、200r/min培养至OD600为0.6-2.0时添加终浓度为0.1-10mmol/L的IPTG,16-37℃,200r/min诱导。
3、测定添加不同前体物质培养后的初始菌株E.coli BL21/pET-21a胞内NAD+含量。结果如表3所示,其中添加了喹啉酸、烟酸及烟酰胺后对胞内辅酶的生成量促进作用较大,相比空白对照组,分别提高了近64%、105%和67%。
表3添加不同前体物质后E.coli BL21/pET-21a胞内NAD+的含量
实施例4
(1)根据实施例3的方法配制功能因子促进剂。
(2)对多基因共表达重组菌株(实施例2制备的重组菌)进行发酵培养,先用LB氨苄青霉素平板活化重组菌株,挑取单菌落接入5mL含有终浓度100μg/mL氨苄青霉素的LB试管中,培养12h后再按1%的接种量转接于50mL的LB发酵培养基中,添加终浓度为1-100mg/L的前体物质,37℃、200r/min培养至OD600为0.6-2.0时添加终浓度为0.1-10mmol/L的IPTG,16-37℃,200r/min诱导。
(3)收集发酵培养后的菌体进行SDS-PAGE分析,组合共表达的关键限速酶酶正常表达后进行胞内NAD+含量的测定,结果如表4所示,添加了前体物质的共表达重组菌株胞内的辅酶含量均有所提高。其中效果最为明显的是E.coli BL21/pET-21a-nadE-pncB和E.coliBL21/pET-21a-pncB-nadE-pncA在添加烟酸后,NAD+含量可达到近32μmol/g DCW。
表4多基因共表达的重组菌株组合不同的前体物质对胞内NAD+含量的影响
本发明配制0~100mg/L的功能因子促进剂,并将其用于重组大肠杆菌的发酵培养,结果显示,均能有效促进胞内NAD+的含量,双基因共表达组合中,喹啉酸对E.coli BL21/pET-21a-nadC-nadD和E.coli BL21/pET-21a-nadC-nadE的促进作用最大,使其胞内氧化型辅酶含量分别达到15.7μmol/g DCW和19.9μmol/g DCW。烟酸对E.coli BL21/pET-21a-pncB-nadE和E.coli BL21/pET-21a-nadE-pncB胞内辅酶含量提高幅度明显,分别可达到30.6μmol/g DCW和32.5μmol/g DCW。烟酰胺对E.coli BL21/pET-21a-pncA-nadE促进作用较大,胞内辅酶含量达24.8μmol/g DCW。
三个基因共表达组合中,烟酰胺对E.coli BL21/pET-21a-nadC-nadD-nadE的提高幅度较大,其他菌株均是烟酸起到了更明显的提高辅酶含量的作用。
所有效果明显的重组菌株与未添加前体物质相比,均提高了56%以上。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!
- 新的乙酰辅酶a羧化酶(acc)抑制剂及其在糖尿病、肥胖症和代谢综合征中的应用的制作方法
- 新的乙酰辅酶a羧化酶(acc)抑制剂及其在糖尿病、肥胖症和代谢综合征中的应用的制作方法
- 用作制备3
- 乙酰辅酶a羧化酶剪接变体及其用途的制作方法
- 新的乙酰辅酶a羧化酶(acc)抑制剂及其在糖尿病、肥胖症和代谢综合征中的应用的制作方法
- 氧肟酸类组蛋白去乙酰化酶抑制剂及其制备方法和用途的制作方法
- 人类病原体艰难梭菌中乙酰辅酶a合成酶的抑制剂的制作方法
- 作为乙酰辅酶a羧化酶(acc)抑制剂的螺苯并二氢吡喃-4-酮的制作方法
- 一种新的多肽——人过氧化物酶体脂肪酸乙酰辅酶a氧化酶46.97和编码这种多肽的多核苷酸的制作方法
- 基于在线控制氧消耗速率的辅酶q10发酵新工艺的制作方法