邻苯二甲腈化合物的制作方法
相关申请的交叉引用
本申请要求基于于2015年9月24日提交的韩国专利申请第10-2015-0135856号的优先权益,其公开内容通过引用整体并入本文。
本申请涉及邻苯二甲腈化合物、邻苯二甲腈树脂、可聚合组合物、预聚物、复合材料、其前体、及其生产方法和用途。
背景技术:
邻苯二甲腈化合物可以应用于多种应用。例如,邻苯二甲腈化合物可以用作所谓的邻苯二甲腈树脂的原料。例如,通过将邻苯二甲腈树脂浸没在填料(例如,玻璃纤维或碳纤维)中而形成的复合材料可以用作汽车、飞机或船舶的材料。用于生产所述复合材料的方法可包括例如以下过程:将邻苯二甲腈和固化剂的混合物或通过所述混合物的反应形成的预聚物与填料混合,然后使混合物固化(参见例如专利文献1)。
邻苯二甲腈化合物的其他用途可包括作为酞菁染料的前体的用途。例如,邻苯二甲腈化合物可与金属配混以用作颜料。
邻苯二甲腈化合物还可以用作荧光增白剂或照相增感剂的前体,或者酸酐的前体等。例如,邻苯二甲腈化合物可以通过适当的氧化过程和脱水过程转化成酸酐,并且这样的酸酐还可以用作聚酰胺酸或聚酰亚胺等的前体。
[现有技术文献]
[专利文献]
(专利文献1)韩国专利第0558158号
技术实现要素:
技术问题
本申请可以提供新的邻苯二甲腈化合物及其用途。作为用途,可以例举邻苯二甲腈树脂、可聚合组合物、预聚物、复合材料、颜料、荧光增白剂、照相增感剂或酸酐的前体或原料。
技术方案
本申请涉及邻苯二甲腈化合物。所述化合物可由下式1表示。
[式1]
在式1中,ar1和ar2各自独立地为经至少一个氨基或羟基取代的芳香族二价基团,l、l1和l2各自独立地为单键、亚烷基、烷叉基、氧原子、硫原子或-s(=o)2-,并且r1至r10各自独立地为氢、烷基、烷氧基、芳基或氰基,前提条件是r1至r5中的至少两个为氰基且r6至r10中的至少两个为氰基。
在此,ar1和ar2可彼此相同或不同,并且l、l1和l2可彼此相同或不同。
在本申请中,除非另有说明,否则术语芳香族二价基团可意指衍生自苯、含有苯的化合物或前述物质的任一种衍生物的二价基团。在此,含有苯的化合物可意指具有以下结构的化合物:两个或更多个苯环在共用两个碳原子的同时各自稠合,或者通过合适的连接基团连接。芳香族二价基团可包含例如6至25、6至20、6至15、或6至12个碳原子。在式1中,为芳香族二价基团的ar1和ar2可经至少1个、1至5、1至4、1至3、或1至2个氨基或羟基取代,并且适当地可经氨基取代。在一些情况下,除了氨基或羟基以外,芳香族二价基团可任选地经一个或更多个取代基取代。
在一个实例中,芳香族二价基团可以是衍生自下式2至4的任一种芳香族化合物的基团。
[式2]
在式2中,r1至r6各自独立地为氢、烷基、烷氧基、芳基、羟基或氨基,前提条件是r1至r6中的至少两个形成自由基,并且r1至r6中的至少一个为羟基或氨基。
[式3]
在式3中,r1至r8各自独立地为氢、烷基、烷氧基、芳基、羟基或氨基,前提条件是r1至r8中的至少两个形成自由基,并且r1至r8中的至少一个为羟基或氨基。
[式4]
在式4中,r1至r10各自独立地为氢、烷基、烷氧基、芳基、羟基或氨基,前提条件是r1至r10中的至少两个形成自由基,l为亚烷基、烷叉基、氧原子或硫原子,并且r1至r10中的至少一个为羟基或氨基。
式2中的r1至r6、式3中的r1至r8或式4中的r1至r10各自独立地为氢、烷基、烷氧基、芳基、羟基或氨基,前提条件是这些中的至少两个形成自由基。在此,形成自由基可意指该位置与式1的另一要素连接。例如,在式1中的ar1的情况下,形成自由基的位置中的任一位置可直接与式1中的l1连接形成共价键,并且其他位置可直接与式1中的l连接形成共价键。在式1中的ar2的情况下,形成自由基的位置中的任一位置可直接与式1中的l2连接形成共价键,并且其他位置可直接与式1中的l连接。不形成自由基的上述各取代基中的至少1个、1至5、1至4、1至3、或1至2个可为氨基或羟基,并且剩余的取代基可为氢、烷基或烷氧基;氢或烷基。在一个实例中,在式2中,r1和r4或r1和r3可形成自由基。在这种情况下,不形成自由基的取代基中的1至3或1至2个可为氨基或羟基,并且其他取代基可各自独立地为氢、烷基、烷氧基或芳基;氢、烷基或烷氧基;或者氢或烷基。此外,在式3中,r1、r6、r7和r8中的任一个以及r2、r3、r4和r5中任一个可形成自由基。在这种情况下,不形成自由基的取代基中的1至5、1至4、1至3、或1至2个可为氨基或羟基,并且其他取代基可各自独立地为氢、烷基、烷氧基或芳基;氢、烷基或烷氧基;或者氢或烷基。此外,在式4中,r1至r5中的任一个和r6至r10中的任一个可形成自由基。在这种情况下,不形成自由基的取代基中的1至5、1至4、1至3、或1至2个可为氨基或羟基,并且其他取代基可各自独立地为氢、烷基、烷氧基或芳基;氢、烷基或烷氧基;或者氢或烷基。此外,在式4中,l可为亚烷基、烷叉基、氧原子或硫原子,并且在另一个实例中,l可为亚烷基、烷叉基或氧原子,或氧原子。
作为式1中的ar1或ar2的一般实例,可以例举式2的芳香族二价基团。在这种情况下,以式2的r1至r6中与式1的l形成共价键的取代基为基准的间位或对位处的取代基可为羟基或氨基。
在本申请中,除非另有说明,否则术语烷基可为具有1至20个碳原子、1至16个碳原子、1至12个碳原子、1至8个碳原子、或1至4个碳原子的烷基。烷基可为线性、支化或环状的,并且可任选地经一个或更多个取代基取代。
在本申请中,除非另有说明,否则术语烷氧基可为具有1至20个碳原子、1至16个碳原子、1至12个碳原子、1至8个碳原子、或1至4个碳原子的烷氧基。烷氧基可为线性、支化或环状的,并且可任选地经一个或更多个取代基取代。
在本申请中,除非另有说明,否则术语芳基可意指衍生自如在芳香族二价基团的项目中描述的苯、包含苯结构的化合物、或其任一种衍生物的一价基团。芳基可包含例如6至25、6至20、6至15、或6至12个碳原子。作为芳基的具体种类,可以例举苯基、苄基、联苯基或萘基等,但不限于此。此外,在本申请中的芳基的范围内,不仅可包括通常被称为芳基的官能团,也可包括所谓的芳烷基或芳基烷基。
在本申请中,除非另有说明,否则术语亚烷基或烷叉基可意指具有1至20个碳原子、1至16个碳原子、1至12个碳原子、1至8个碳原子、或1至4个碳原子的亚烷基或烷叉基。亚烷基或烷叉基可为线性、支化或环状的。此外,亚烷基或烷叉基可任选地经一个或更多个取代基取代。
作为可任选地取代本申请中的烷基、烷氧基、芳基、芳香族二价基团、亚烷基或烷叉基的取代基,可以例举卤素,例如氯或氟;环氧基,例如缩水甘油基、环氧烷基、环氧丙氧基烷基或脂环族环氧基;丙烯酰基;甲基丙烯酰基;异氰酸酯基;硫醇基;烷基;烷氧基;或芳基,但不限于此。
在式1中,l、l1和l2可为单键、亚烷基、烷叉基、氧原子、硫原子或-s(=o)2-。
在一个实例中,式1的l可为单键、亚烷基或烷叉基、或-s(=o)2-。亚烷基或烷叉基可任选地经至少一个卤素原子或卤代烷基取代,即,经卤素原子取代的烷基,并且在一些情况下,可任选地经除卤素原子以外的取代基取代。此外,上文中的术语单键是在相应位置不存在分隔原子的情况,例如,如果l为单键,则可以得到其中ar1和ar2直接连接的结构。
在式1中,l1和l2可为亚烷基、烷叉基或氧原子,并且在一个实例中,l1和l2可为氧原子。
在式1中,r1至r10各自独立地为氢、烷基、烷氧基、芳基或氰基,前提条件是r1至r5中的至少两个为氰基且r6至r10中的至少两个为氰基。在另一个实例中,不为氰基的r1至r10可各自独立地为氢、烷基或烷氧基;或者氢或烷基。在一个实例中,式1中的r2至r4中的任意两个和r7至r9中的任意两个可为氰基,并且剩余的取代基可各自独立地为氢、烷基、烷氧基或芳基;氢、烷基或烷氧基;或者氢或烷基。
式1的化合物可以有效地用于其中已知所谓的邻苯二甲腈化合物可适用的各种应用中。例如,邻苯二甲腈化合物可以有效地用作能够生产所谓的邻苯二甲腈树脂的原料或前体。所述化合物可表现出低的熔融温度,与固化剂具有优异的反应性,并且表现出宽的加工窗口以有效地应用于应用。除了上述应用以外,所述化合物可用作染料(例如,酞菁染料)的前体,或者荧光增白剂、照相增感剂或酸酐的前体或原料等。
式1的化合物可以通过有机化合物的已知合成方法来合成。例如,式1的化合物可以通过使经氨基或羟基取代、同时具有酚羟基的芳香族化合物与具有至少2个氰基的芳香族化合物反应的方法(例如,硝基置换法)等来合成。在有机化学领域中,能够形成式1的化合物的结构的芳香族化合物是已知的,并且考虑到期望的结构,这样的化合物可以全部应用于上述化合物的生产。
本申请还涉及所述化合物的用途。作为所述化合物的用途,可以例举邻苯二甲腈树脂、酞菁染料、荧光增白剂、照相增感剂或酸酐的原料或前体,如上所述。
作为用途的一个实例,例如,本申请可涉及邻苯二甲腈树脂。邻苯二甲腈树脂可包含衍生自式1的化合物的聚合单元。在本申请中,术语衍生自某种化合物的聚合单元可意指通过所述化合物的聚合或固化而形成的聚合物的骨架。
在邻苯二甲腈树脂中,式1的化合物的聚合单元可为通过如下反应形成的聚合单元:上述化合物与固化剂的反应、式1的化合物之间的反应、或者式1的化合物与另一种邻苯二甲腈化合物的反应。
式1的化合物可包含与氰基自主反应形成聚合单元而不需要任何单独的固化剂的氨基或羟基。因此,邻苯二甲腈树脂可以仅由式1的化合物形成,并且如果需要的话,式1的化合物可以用作其他邻苯二甲腈单体或包含其的组合物的固化剂。在这种情况下,由于式1的化合物本身参与反应并且成为邻苯二甲腈树脂的组分,故可以提高固化速率和固化密度,并且即使用作固化剂的式1的化合物的含量增加,物理特性也不劣化。
当通过仅使用式1的化合物或者通过将除式1的化合物以外的邻苯二甲腈与式1的化合物一起应用来形成邻苯二甲腈树脂时,如上所述不需要固化剂,但是在一些情况下,也可混合和使用其他合适的固化剂。
此外,除了式1的化合物的聚合单元以外,邻苯二甲腈树脂还可包含其他邻苯二甲腈化合物的聚合单元。在这种情况下,能够选择和使用的邻苯二甲腈化合物的种类没有特别限制,并且可以应用已知可用于形成邻苯二甲腈树脂并控制其物理特性的已知化合物。作为这样的化合物的实例,可以例举美国专利第4,408,035、5,003,039、5,003,078、5,004,801、5,132,396、5,139,054、5,208,318、5,237,045、5,292,854、或5,350,828号中公开的化合物,但不限于此。
如上所述,如果需要的话,可将固化剂与式1的化合物一起使用,其中固化剂的种类没有特别限制,只要其通常用于形成邻苯二甲腈树脂即可。这样的固化剂在包括上述美国专利的各种文献中是已知的。
在一个实例中,胺化合物(例如,芳香族胺化合物)或羟基化合物可以用作固化剂。在本申请中,羟基化合物可意指在分子中含有至少一个或两个羟基的化合物。
本申请还涉及可聚合组合物。所述可聚合组合物可包含式1的化合物。所述可聚合组合物能够形成所谓的邻苯二甲腈树脂,并且主要包含式1的化合物,并且还可包含或不包含固化剂。即,如上所述,式1的化合物可以自主地进行固化反应,并因此可聚合组合物可不包含用作固化剂的胺化合物或羟基化合物。即,除了式1的化合物以外,可聚合组合物可不包含具有胺基或羟基的化合物。
如果需要的话,可包含适当的固化剂,其中作为包含在可聚合组合物中的固化剂,例如,可使用如上所述的固化剂。当包含固化剂时,可聚合组合物中的固化剂的比例没有特别限制。例如,考虑到包含在组合物中的可固化组分(例如,式1的化合物)的比例或种类,可以调节所述比例,使得可以确保期望的可固化性。例如,可以以每摩尔包含在可聚合组合物中的式1的化合物约0.02摩尔至2摩尔或0.02摩尔至1.5摩尔的量包含固化剂。然而,上述比例仅是本申请的实例。通常,如果可聚合组合物中的固化剂的比例较高,则加工窗口倾向于变窄;而如果固化剂的比例较低,则可固化性倾向于变得不足,因此可以考虑到这一点来选择合适的固化剂比例。
在另一个实例中,可聚合组合物可包含除式1的化合物以外的邻苯二甲腈化合物。如上所述,在上述情况下,式1的化合物可用作固化剂。在这种情况下,即使当用作固化剂的式1的化合物的含量增加时,邻苯二甲腈树脂的含量也不降低,并且可以获得高固化度的树脂。当作为固化剂的式1的化合物与其他邻苯二甲腈化合物一起包含在可聚合组合物中时,其比例没有特别限制,并且其可以以例如2摩尔%至95摩尔%包含在整个组合物中。
包含式1的化合物的可聚合组合物即使在低温,例如最高约350℃的温度下也可以快速且容易地固化,并且可以表现出低的熔融温度。此外,当如上所述包含式1的化合物作为固化剂并且其比例变得过大时,式1的化合物参与反应并成为最终产物(邻苯二甲腈树脂等)的成分,因此可以防止物理特性劣化。
除了式1的化合物以外,可聚合组合物还可包含各种添加剂,包括其他邻苯二甲腈化合物等。作为这样的添加剂的实例,可以例举各种填料。可以用作填料的材料的种类没有特别限制,并且可根据预期用途使用任何已知的合适填料。作为示例性填料,可以例举金属材料、陶瓷材料、玻璃、金属氧化物、金属氮化物或基于碳的材料等,但不限于此。此外,填料的类型也没有特别限制并且可以是各种形式,例如纤维材料,如芳族聚酰胺纤维、玻璃纤维、碳纤维或陶瓷纤维;或者由所述材料形成的织造织物、非织造织物、线或绳;包含纳米颗粒的微粒;多边形或其他无定形形式。在此,作为基于碳的材料,可以例举石墨、石墨烯或碳纳米管等,或者其衍生物或异构体例如氧化物等。然而,可聚合组合物还可包含的组分不限于上述物质,并且根据用途,还可包含已知可应用于生产所谓的工程塑料(例如聚酰亚胺、聚酰胺或聚苯乙烯)的各种单体或其他添加剂而没有限制。
本申请还涉及通过可聚合组合物,即,包含式1的化合物的可聚合组合物的反应而形成的预聚物。
在本申请中,术语预聚物状态可意指这样的状态:其中可聚合组合物中的式1的化合物和固化剂处于以一定程度聚合的状态(例如,发生所谓的a阶段或b步骤的聚合的状态),而没有达到完全聚合的状态,并且表现出适当的流动性,例如,允许加工下文将描述的复合材料。
如上所述,预聚物还可表现出优异的可固化性和低的熔融温度。
除了上述组分以外,预聚物还可包含任何已知的添加剂。作为这样的添加剂的实例,可以例举上述填料等,但不限于此。
本申请还涉及复合材料。所述复合材料可包含上述邻苯二甲腈树脂和填料。如上所述,可以通过本申请的式1的化合物来实现优异的可固化性、低的熔融温度和宽的加工窗口,并且因此,可以容易地形成包含各种填料的具有优异的物理特性的所谓的增强树脂复合材料(增强聚合物复合材料)。由此形成的复合材料可包含邻苯二甲腈树脂和填料,并且可应用于例如各种应用,包括耐用品例如汽车、飞机或船舶等。
填料的种类没有特别限制并且可以考虑到预期用途而适当地选择。可用填料的具体类型如上所述。
此外,填料的比例没有特别限制,并且可根据预期用途而设置在适当的范围内。
本申请还涉及用于生产复合材料的前体,其中所述前体可包含例如如上所述的可聚合组合物和填料,或如上所述的预聚物和填料。
复合材料可以使用前体以已知的方式制备。例如,复合材料可以通过使前体固化来形成。
在一个实例中,前体可通过将可聚合组合物或预聚物与通过加热等处于熔融的状态的填料共混来制备,所述可聚合组合物通过将上述式1的化合物与处于熔融状态的固化剂配混来制备。例如,上述复合材料可以通过将由此制备的前体模制成期望的形状,然后使其固化来制备。可聚合组合物或预聚物具有低的熔融温度和宽的加工温度以及优异的可固化性,因此可以在上述过程中有效地进行模制和固化。
在上述过程中,用于形成预聚物等的方法,通过将这样的预聚物与填料配混并对其进行加工和使其固化来生产复合材料的方法等可根据已知的方法进行。
本申请还可涉及包含所述化合物的酞菁染料的前体、荧光增白剂的前体、或照相增感剂的前体,或衍生自所述化合物的酸酐。使用所述化合物来形成前体的方法或生产酸酐的方法没有特别限制,并且可以应用能够使用邻苯二甲腈化合物生产所述前体或酸酐的所有已知方法。
有益效果
本申请可以提供邻苯二甲腈化合物及其用途,所述邻苯二甲腈化合物可以自主固化以形成邻苯二甲腈树脂,或者可以与其他邻苯二甲腈化合物组合以用作固化剂。
邻苯二甲腈化合物即使在低温下也可以以高速度自固化以形成邻苯二甲腈树脂,并且不会由于使用现有的固化剂而引起缺陷。
此外,邻苯二甲腈化合物可以通过与另一种化合物共混而用作固化剂,并且在这种情况下,即使当用作固化剂的化合物的含量增加时,整个邻苯二甲腈树脂的含量也不降低,由此可以提供优异固化度的树脂。
附图说明
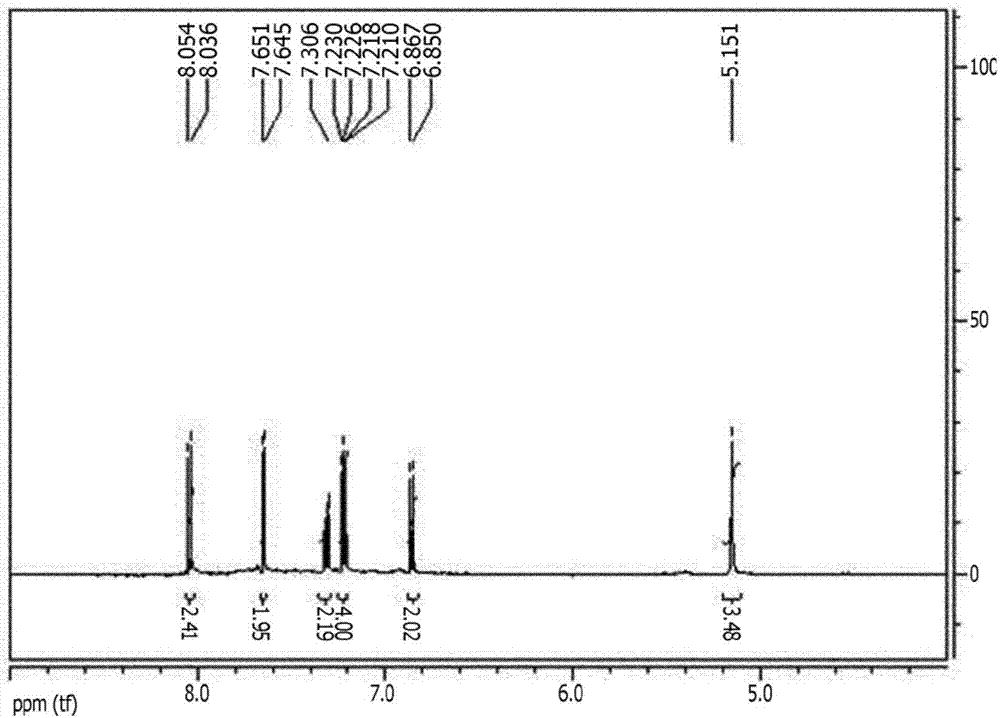
图1至6各自是在制备例1至6中制备的化合物的nmr分析结果。
具体实施方式
将通过实施例和比较例具体描述本申请的邻苯二甲腈树脂等,但是树脂等的范围不限于以下实施例。
1.核磁共振(nmr)分析
使用来自agilent的500mhznmr仪器根据制造商手册进行nmr分析。通过将化合物溶解在dmso(二甲基亚砜)-d6中来制备用于nmr测量的样品。
2.dsc(差示扫描量热法)分析
用来自tainstrument的q20系统在以10℃/分钟的升温速率将温度从35℃升高至450℃的同时,在n2流气氛中进行dsc分析。
3.tga(热重分析)分析
使用来自mettler-toledo的tgae850仪器进行tga分析。在制备例中制备的化合物的情况下,在以10℃/分钟的升温速率将温度从25℃升高至800℃的同时,在n2流气氛下对其进行分析。
制备例1.化合物(pn1)的合成
以以下方式合成下式a的化合物(pn1)。将54.9g下式b的化合物和150gdmf(二甲基甲酰胺)放入3颈rbf(圆底烧瓶)中,在室温下搅拌并溶解。随后,向其中添加51.9g下式c的化合物,并向其中添加50gdmf,然后搅拌混合物并使其溶解。随后,将62.2g碳酸钾和50gdmf一起添加到其中,并使混合物在搅拌下在将温度升高至85℃的状态下反应,然后冷却至室温。将冷却的反应溶液倒入0.2n盐酸水溶液中以中和并沉淀,随后过滤,然后用水洗涤。其后,将过滤的反应物在真空烘箱中于100℃下干燥1天,并且在除去水和残留溶剂之后,以约85重量%的收率获得下式a的化合物。式a的化合物的nmr结果描述于图1中。
[式a]
[式b]
[式c]
制备例2.化合物(pn2)的合成
以以下方式合成下式d的化合物(pn2)。将32.4g下式e的化合物和130gdmf(二甲基甲酰胺)放入3颈rbf(圆底烧瓶)中,在室温下搅拌并溶解。随后,向其中添加51.9g以上制备例1中的式c的化合物,并向其中添加50gdmf,然后搅拌混合物并使其溶解。随后,将62.2g碳酸钾和50gdmf一起添加到其中,并在搅拌下将温度升高至85℃。使混合物在该状态下反应约5小时,然后冷却至室温。将冷却的反应溶液倒入0.2n盐酸水溶液中以中和并沉淀,随后过滤,然后用水洗涤。其后,将过滤的反应物在真空烘箱中于100℃下干燥1天,并且在除去水和残留溶剂之后,以约80重量%的收率获得下式d的化合物。式d的化合物的nmr结果描述于图2中。
[式d]
[式e]
制备例3.化合物(pn3)的合成
以以下方式合成下式f的化合物(pn3)。将42g下式g的化合物和200gdmf(二甲基甲酰胺)放入3颈rbf(圆底烧瓶)中,在室温下搅拌并溶解。随后,向其中添加51.9g以上制备例1中的式c的化合物,并向其中添加50gdmf,然后搅拌混合物并使其溶解。随后,将62.2g碳酸钾和50gdmf一起添加到其中,并在搅拌下将温度升高至85℃。使混合物在该状态下反应约5小时,然后冷却至室温。将冷却的反应溶液倒入0.2n盐酸水溶液中以中和并沉淀,随后过滤,然后用水洗涤。其后,将过滤的反应物在真空烘箱中于100℃下干燥1天,并且在除去水和残留溶剂之后,以约82重量%的收率获得下式f的化合物。式f的化合物的nmr结果描述于图3中。
[式f]
[式g]
制备例4.化合物(pn4)的合成
以以下方式合成下式h的化合物(pn4)。将27.9g下式i的化合物和100gdmf(二甲基甲酰胺)放入3颈rbf(圆底烧瓶)中,在室温下搅拌并溶解。随后,向其中添加51.9g以上制备例1中的式c的化合物,并向其中添加50gdmf,然后搅拌混合物并使其溶解。随后,将62.2g碳酸钾和50gdmf一起添加到其中,并在搅拌下将温度升高至85℃。使混合物在该状态下反应约5小时,然后冷却至室温。将冷却的反应溶液倒入0.2n盐酸水溶液中以中和并沉淀,随后过滤,然后用水洗涤。其后,将过滤的反应物在真空烘箱中于100℃下干燥1天,并且在除去水和残留溶剂之后,以约83重量%的收率获得下式h的化合物。式h的化合物的nmr结果描述于图4中。
[式h]
[式i]
制备例5.化合物(pn5)的合成
将50.4g下式k的化合物和150gdmf(二甲基甲酰胺)放入3颈rbf(圆底烧瓶)中,在室温下搅拌并溶解。随后,向其中添加51.9g以上制备例1中的式c的化合物,并向其中添加50gdmf,然后搅拌混合物并使其溶解。随后,将62.2g碳酸钾和50gdmf一起添加到其中,并在搅拌下将温度升高至85℃。使混合物在该状态下反应约5小时,然后冷却至室温。将冷却的反应溶液倒入0.2n盐酸水溶液中以中和并沉淀,随后过滤,然后用水洗涤。其后,将过滤的反应物在真空烘箱中于100℃下干燥1天,并且在除去水和残留溶剂之后,以约87重量%的收率获得下式j的化合物(pn5)。式j的化合物的nmr结果描述于图5中。
[式j]
[式k]
制备例6.化合物(pn6)的合成
以以下方式合成下式l的化合物。将32.7g下式m的化合物和120gdmf(二甲基甲酰胺)放入3颈rbf(圆底烧瓶)中,在室温下搅拌并溶解。随后,向其中添加51.9g以上制备例1中的式c的化合物,并向其中添加50gdmf,然后搅拌混合物并使其溶解。随后,将62.2g碳酸钾和50gdmf一起添加到其中,并在搅拌下将温度升高至85℃。使混合物在该状态下反应约5小时,然后冷却至室温。将冷却的反应溶液倒入0.2n盐酸水溶液中以中和并沉淀,随后过滤,然后用水洗涤。其后,将过滤的反应物在真空烘箱中于100℃下干燥1天,并且在除去水和残留溶剂之后,以约80重量%的收率获得下式l的化合物(pn6)。式l的化合物的nmr结果描述于图5中。
[式l]
[式m]
制备例7.化合物(ca)的合成
作为下式n的化合物(ca),购买并使用来自tci(tokyochemicalindustryco.,ltd.)的商业产品而无需进一步纯化。
[式n]
实施例1.
通过dsc分析确定制备例1的化合物(pn1)的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使制备例1的化合物(pn1)在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
实施例2.
通过dsc分析确定制备例2的化合物(pn2)的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使制备例2的化合物(pn2)在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
实施例3.
通过dsc分析确定制备例3的化合物(pn3)的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使制备例3的化合物(pn3)在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
实施例4.
将制备例1的化合物(pn1)和制备例4的化合物(pn4)混合,使得制备例1的化合物(pn1)以每摩尔制备例4的化合物(pn4)约0.2摩尔存在。然后,通过dsc分析确定混合物的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使混合物在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
实施例5.
将制备例1的化合物(pn1)和制备例5的化合物(pn5)混合,使得制备例1的化合物(pn1)以每摩尔制备例5的化合物(pn5)约0.2摩尔存在。然后,通过dsc分析确定混合物的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使混合物在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
实施例6.
将制备例1的化合物(pn1)和制备例6的化合物(pn6)混合,使得制备例1的化合物(pn1)以每摩尔制备例6的化合物(pn6)约0.2摩尔存在。然后,通过dsc分析确定混合物的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使混合物在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
比较例1
通过dsc分析确定制备例4的化合物(pn4)的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使制备例4的化合物(pn4)在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
比较例2
通过dsc分析确定制备例5的化合物(pn5)的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使制备例5的化合物(pn5)在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
比较例3
通过dsc分析确定制备例6的化合物(pn6)的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使制备例6的化合物(pn6)在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
比较例4
将制备例4的化合物(pn4)和制备例7的化合物(ca)混合,使得制备例7的化合物(ca)以每摩尔制备例4的化合物(pn4)约0.2摩尔存在。然后,通过dsc分析确定混合物的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使混合物在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
比较例5
将制备例5的化合物(pn5)和制备例7的化合物(ca)混合,使得制备例7的化合物(ca)以每摩尔制备例5的化合物(pn5)约0.2摩尔存在。然后,通过dsc分析确定混合物的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使混合物在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
比较例6
将制备例6的化合物(pn6)和制备例7的化合物(ca)混合,使得制备例7的化合物(ca)以每摩尔制备例6的化合物(pn6)约0.2摩尔存在。然后,通过dsc分析确定混合物的放热开始温度和放热最高温度。此外,通过对其中使用ir固化炉使混合物在240℃下固化2小时的材料进行tga分析确定在td10%和800℃下的残余物。
上述实施例和比较例的测量结果概述并描述于下表1中。
[表1]
在表1中,首先,将实施例1至3和比较例1至3的结果进行比较,可以确认在实施例1至3的情况下的固化峰,即使各自处于单一化合物的状态下,因此可以确定放热开始温度和放热最高温度;而在比较例1至3中,即使在温度升高至450℃的状态下也不能确认固化峰。此外,在实施例1至3的情况下,作为在ir固化炉中固化的结果,即使在相对短的时间(2小时)内也发生交联反应,因此在800℃下的残余物为60%至70%,并且表现出高的耐热性,但是在比较例1至3的情况下,即使在保持在固化炉中之后,在800℃下的残余物也处于6%至20%的水平。
在实施例4至6(其分别为比较例1至3的化合物和实施例1的化合物的混合物)的情况下,从确认dsc的结果可以看出,发生了其中固化反应不单独进行的比较例1至3的化合物的固化,由此在800℃下的残余物大大提高至60%至70%。
另一方面,将比较例4至6和实施例4至6的结果进行比较,可以确认,与应用作为已知固化剂的制备例7的化合物(ca)的比较例4至6相比,应用制备例1的化合物(pn1)的实施例4至6中的固化在较低的温度下开始,由此可以看出,实施例4至6表现出更优异的可快速固化性。
从这些结果可以看出,通过使用本发明的化合物,可以确保可自固化性,从而避免在引入单分子固化剂等时发生的空隙问题等。此外,由上述特性,可以通过防止因固化剂含量引起的单体比例降低等来确保更高的固化度,从而即使在热和机械特性方面也预期改善的结果,并且通过经由降低固化温度和缩短固化时间而缩短加工时间来提高生产率。
- 还没有人留言评论。精彩留言会获得点赞!