用作药物、特别是用于治疗帕金森氏病的新型双环化合物的制作方法
背景技术:
在过去的50年中,儿茶酚-O-甲基转移酶(COMT),一种参与大脑中神经递质降解的镁依赖性酶,已成为治疗各种外周和中枢神经系统疾病的有吸引力的靶点。该酶以两种形式存在:可溶形式(S-COMT)和膜结合形式(MB-COMT),并且广泛表达。
COMT抑制剂与多巴胺前体左旋多巴(L-DOPA)和芳香族氨基酸脱羧酶(AADC)抑制剂的联合施用增加了L-DOPA在体内的半衰期。因此,COMT抑制剂是帕金森氏病辅助治疗的候选药物。临床相关的COMT抑制剂例如有硝替卡朋(OR-462)、恩他卡朋(OR-611)、托卡朋(Ro 40-7592)、硝苄卡朋(BIA3-202)和奥比卡朋(BIA9-1967)。迄今为止COMT抑制剂的唯一临床应用是在III期试验中使用恩他卡朋和奥比卡朋与L-DOPA和AADC抑制剂联合施用。由于无有效性或安全性问题,其他抑制剂已停产或退出市场。
在多种情况下,作用于中枢和外周的托卡朋的使用导致严重的肝毒性。主要作用于外周的恩他卡朋具有更有利的毒性特征,但存在生物利用度问题。
本发明的目的在于提供具有减少的副作用(肝毒性)和增加的效力的新型COMT抑制剂。此目的通过独立权利要求的主题而实现。
术语和定义
如本文所用,除非另有说明,否则术语“经取代的”在化学部分的语境中使用时表示该化学部分的一个或多个氢原子已经被选自以下的非氢取代基原子或取代基部分所替代:-D(氘)、-F、-Cl、-Br、-I、=O、-OH、-OR、=S、-SH、-OSO3H、-OSO3R、-SO3H、-SO2R、-SO2NH2、-SO2NHR、-SO2NR2、-OPO32、-NH2、-NHR、-COOH、-CONH2、-OCOOH、-NHCONH2、-CN、-NC、-CNO、-NCO、-CNS、-SCN、CF3、CHF2、CH2F,其中,R是C1-C8烷基、C2-C8烯基、C2-C8炔基、C3-C10环烷基、5至10元芳基或杂芳基,以及由2至25个选自C、N、O、S和F的原子加任何适当量的H原子组成的取代基部分,其中,取代基部分的分子量为29g/mol~400g/mol。
如本文所用,除非另有说明,否则术语“烷基”是指直链或支链的饱和烃取代基。烷基的实例包括但不限于甲基、乙基、丙基、正丁基、异丁基、戊基、异戊基、新戊基、己基、2-甲基戊基、3-甲基戊基、2,2-甲基戊基、2,3-甲基戊基等。
如本文所用,除非另有说明,否则术语“烯基”是指包含至少一个碳-碳双键的不饱和烃取代基。烯基可以是直链或支链的。
如本文所用,除非另有说明,否则术语“炔基”是指包含至少一个碳-碳三键的不饱和烃取代基。炔基可以是直链或支链的。一个实例是乙炔基(CCH)。
如本文所用,除非另有说明,否则术语“杂烷基”是指直链或支链的饱和烃取代基,其中,至少一个碳被氧原子、硫原子或氮原子代替。
如本文所用,除非另有说明,否则术语“环烷基”是指饱和的环状烃取代基。
如本文所用,除非另有说明,否则术语“杂环烷基”是指其中至少一个碳被氧、氮或硫原子代替的饱和环状基团。饱和杂环取代基的实例包括但不限于吗啉、哌嗪、吡咯烷、四氢噻吩、四氢呋喃、咪唑烷、吡唑烷、噁唑烷、噻唑烷、哌啶、噁烷基等。
如本文所用,除非另有说明,否则术语“芳基”是指环状芳族烃取代基。
术语“杂芳基”是指其中至少一个碳原子被氧、氮或硫原子代替的芳基化合物。芳基或杂芳基取代基的实例包括但不限于苯、吡啶、吡咯、喹诺酮、咪唑、三嗪、吡嗪、嘧啶、哒嗪、噻嗪、二恶英、四唑、三唑、噻二唑、吡唑、噁唑、噻唑、呋喃等。
技术实现要素:
根据本发明的第一方面,提供了由通式(A)表示的化合物:
其中,
R1选自-OH、-SH和-NH2;
R2选自-NO2、-SO3H、-SO2-OR11、-SO2-NH2、-SO2-NHR11、-SO2-NR112、-PO3H、-PO2-OR11、-CN、-COR11、-CO2R11、-CONHR11、-CONR112、-CF3和卤素;
R3选自H、-NO2、-SO3H、-SO2-OR11、-SO2-NH2、-SO2-NHR11、-SO2-NR112、-PO3H、-PO2-OR11、-CN、-COR11、-CO2R11、-CONHR11、-CONR112、-CF3和卤素;
R4和R5独立地选自-H、经取代的C1-C6烷基、未经取代的C1-C6烷基、经取代的或未经取代的C2-C6烯基、经取代的或未经取代的C3-C6环烷基、卤素、-CN和-NO2;
R6和R7中的一个选自-CONR9R11、-CONR92、-COR9、-COOR9、-SO2-OH、-SO2R9、-SO2-OR9、-SO2-OR11、-SO2NHR11、-SO2NR9R11、-NO2、-CN、-CF3和-SO2NR92;
R6和R7中的另一个选自H、-CONH2、COOH、-SO2-OH、-SO2NH2、-Y-R9、-Y-R92和-R9,
其中,
R9是R10-(Y-R10)n
Y选自-CONR11-、-CO-、-COO-、-SO3-、-SO2NH-、-NR11-、-O-、-NR11CO-、-OCO-、-NR11COO-、-NR11CONR11-、-OCONR11-和-OCOO-
n是选自0、1、2、3和4的整数,
每个R10,独立于任何其他R10,选自经取代的或未经取代的C1-C8烷基、经取代的或未经取代的C2-C8烯基、经取代的或未经取代的C2-C8炔基、经取代的或未经取代的C3-C10环烷基和经取代的或未经取代的5至10元芳基或杂芳基;且
每个R11,独立于任何其他R11,是H或未经取代的C1-C6烷基。
在某些实施方式中,R7是选自以下的电负性吸电子基团:-CONR11R9、-CONR92、-COR9、-COOR9、-SO2-OH、-SO2R9、-SO2-OR9、-SO2NH2、-SO2-NHR9、-SO2NR11R9和-SO2NR92,并且R9在分子“右边”部分上赋予空间位阻,如式所示,因此可以自由地采取上述R9所定义的任何形式。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,并且R4和R5是H。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,并且R4和R5是H,且R6选自H、-CONH2和-CONR92,其中,R9具有与上述定义相同的含义。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,并且R4和R5是H,且R7选自-CONR92和-CONR11R9,其中,R9和R11具有与上述定义相同的含义。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,并且R4和R5是H,R6选自H、-CONH2和-CONR92,且R7选自-CONR92和-CONR11R9,其中,R9和R11具有与上述定义相同的含义。
在某些实施方式中,n是0。
在某些实施方式中,n是1。
在某些实施方式中,n是2。
在某些实施方式中,R6是-CONR11R11,且R7是-CONR9R11,其中,每个R11,独立于任何其他R11,是H或未经取代的C1-C4烷基,并且R9具有与上述定义相同的含义。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,并且R4和R5是H,R6是-CONR11R11,且R7是-CONR9R11,其中,每个R11,独立于任何其他R11,是H或未经取代的C1-C4烷基,并且R9具有与上述定义相同的含义。
在某些实施方式中,R6和R7是-CONR9R11,并且
R9是R11-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,并且R4和R5是H,R6和R7是-CONR9R11,并且
R9是R11-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
在某些实施方式中,R6是-CONR112且R7是-CONR9R11,并且
R9是R11-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,R4和R5是H,R6是-CONR112且R7是-CONR9R11,并且
R9是R11-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
在某些实施方式中,R6是H且R7是-CONR9R11,并且
R9是R11-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,R4和R5是H,R6是H且R7是-CONR9R11,并且
R9是R11-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
在某些实施方式中,R7是-CONR9R11,并且
R9是(CH2)4-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的甲基或乙基。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,R4和R5是H,R6是H且R7是-CONR9R11,并且
R9是(CH2)4-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的甲基或乙基。
在某些实施方式中,R1是-OH,R2是-NO2且R3是H,R4和R5是H,R6是-CONEt2且R7是-CONR9R11,并且
R9是(CH2)4-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的甲基或乙基。
在某些实施方式中,提供的化合物选自:
N2,N2,N3,N3-四乙基-6,7-二羟基-5-硝基-萘-2,3-二甲酰胺(A1)
(乙酸3-[[3-(二乙基氨基甲酰基)-6,7-二羟基-5-硝基-萘-2-羰基]-乙基-氨基]丙基酯)(A2)
N2-(6-氨基己基)-N2,N3,N3-三乙基-6,7-二羟基-5-硝基-萘-2,3-二甲酰胺(A4)
N2,N3,N3-三乙基-6,7-二羟基-N2-[4-[甲基(戊酰基)氨基]丁基]-5-硝基-萘-2,3-二甲酰胺(A5)
N-[6-[[3-(二乙基氨基甲酰基)-6,7-二羟基-5-硝基-萘-2-羰基]-乙基-氨基]己基]氨基甲酸叔丁酯(A6)
4-[4-[[3-(二乙基氨基甲酰基)-6,7-二羟基-5-硝基-萘-2-羰基]-乙基-氨基]丁基-甲基-氨基]-4-氧代-丁酸(A8)
N2-[4-[4-氨基丁酰基(甲基)氨基]丁基]-N2,N3,N3-三乙基-6,7-二羟基-5-硝基-萘-2,3-二甲酰胺(A9)
N-[4-[4-[[3-(二乙基氨基甲酰基)-6,7-二羟基-5-硝基-萘-2-羰基]-乙基-氨基]丁基-甲基-氨基]-4-氧代-丁基]氨基甲酸叔丁酯(A10)
N2,N3,N3-三乙基-6,7-二羟基-N2-[4-[[6-(2-甲氧基苯基)-6-氧代-己酰基]-甲基-氨基]丁基]-5-硝基-萘-2,3-二甲酰胺(A11)
N-[1-环己基-2-[4-[[3-(二乙基氨基甲酰基)-6,7-二羟基-5-硝基-萘-2-羰基]-乙基-氨基]丁基-甲基-氨基]-2-氧代-乙基]氨基甲酸叔丁酯(A13)
根据本发明的另一方面,提供以通式(B)表示的化合物,
其中,
R8选自C、S、N、O、NR11和CR11;
R1、R2、R3、R9和R11具有与上述定义相同的含义。
在某些实施方式中,
R1是-OH,
R2是-NO2且R3是H,并且
R8是S;
并且R9具有与上述定义相同的含义。
某些实施方式由式(B2)、(B4)、(B5)、(B6)、(B8)、(B9)、(B10)、(B11)或(B13)中的任何一个表示。
根据本发明的另一方面,提供了用作药物的根据本发明的上述公开方面和实施方式中任一个的化合物。
根据本发明的另一方面,提供了用作用于预防或治疗选自包含帕金森氏病、阿尔兹海默症、多重耐药性结核病和肥胖相关疾病的组中的病症的药物的根据本发明的上述公开方面和实施方式中任一个的化合物。
根据本发明的另一方面,提供了用于预防或治疗选自包含帕金森氏病、阿尔兹海默症、多重耐药性结核病和肥胖相关疾病的组中的病症的包含根据本发明的上述公开方面和实施方式中任一个的化合物的剂型;其中,所述剂型适用于吸入或口服施用,特别是所述剂型是片剂、胶囊、锭剂、粉末剂、溶液、悬浮液或糖浆。
根据本发明的另一方面,提供用于预防或治疗选自包含帕金森氏病、阿尔兹海默症、多重耐药性结核病和肥胖相关疾病的组中的病症的COMT抑制剂,其特征在于所述抑制剂不与HIBCH结合。
本发明的其他方面和实施方式在以下条款中公开:
条款1:由通式(B)表示的化合物
其中,
R1选自-OH、-SH和-NH2;
R2选自-NO2、-SO3H、-SO2-OR11、-SO2-NH2、-SO2-NHR11、-SO2-NR112、-PO3H、-PO2-OR11、-CN、-COR11、-CO2R11、-CONHR11、-CONR112、-CF3和卤素;
R3选自H、-NO2、-SO3H、-SO2-OR11、-SO2-NH2、-SO2-NHR11、-SO2-NR112、-PO3H、-PO2-OR11、-CN、-COR11、-CO2R11、-CONHR11、-CONR112、-CF3和卤素;
R8选自C、S、N、O、NR11和CR11;
R9是R10-(Y-R10)n
Y选自-CONR11-、-CO-、-COO-、-SO3-、-SO2NH-、-NR11-、-O-、-NR11CO-、-OCO-、-NR11COO-、-NR11CONR11-、-OCONR11-和-OCOO-
n是选自0、1、2、3和4的整数,
每个R10,独立于任何其他R10,选自经取代的或未经取代的C1-C8烷基、经取代的或未经取代的C2-C8烯基、经取代的或未经取代的C2-C8炔基、经取代的或未经取代的C3-C10环烷基和经取代的或未经取代的5至10元芳基或杂芳基;每个R11,独立于任何其他R11,是H或未经取代的C1-C6烷基。
条款2:如条款1所述的由通式(B)表示的化合物,其中,
R1是-OH,
R2是-NO2且R3是H,并且
R8是S;
并且R9具有与条款1中的定义相同的含义。
条款3:如条款1或2所述的化合物,其中n是0。
条款4:如条款1或2所述的化合物,其中n是1。
条款5:如条款1或2所述的化合物,其中n是2。
条款6:如条款1或2所述的化合物,其中n是3。
条款7:如条款1至6中任一项所述的化合物,其中
R9是R11-(Y-R10)n
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的C1、C2、C3或C4烷基。
条款8:如条款1至7中任一项所述的化合物,其中,
R9是(CH2)4-(Y-R10)n,
Y选自-CONR11-、-NR11CO-、-NR11COO-和-NR11CONR11-,
n是1或2,
R10是未经取代的或单取代的C1、C2、C3或C4烷基,
R11是未经取代的甲基或乙基。
其他实施方式为下式:
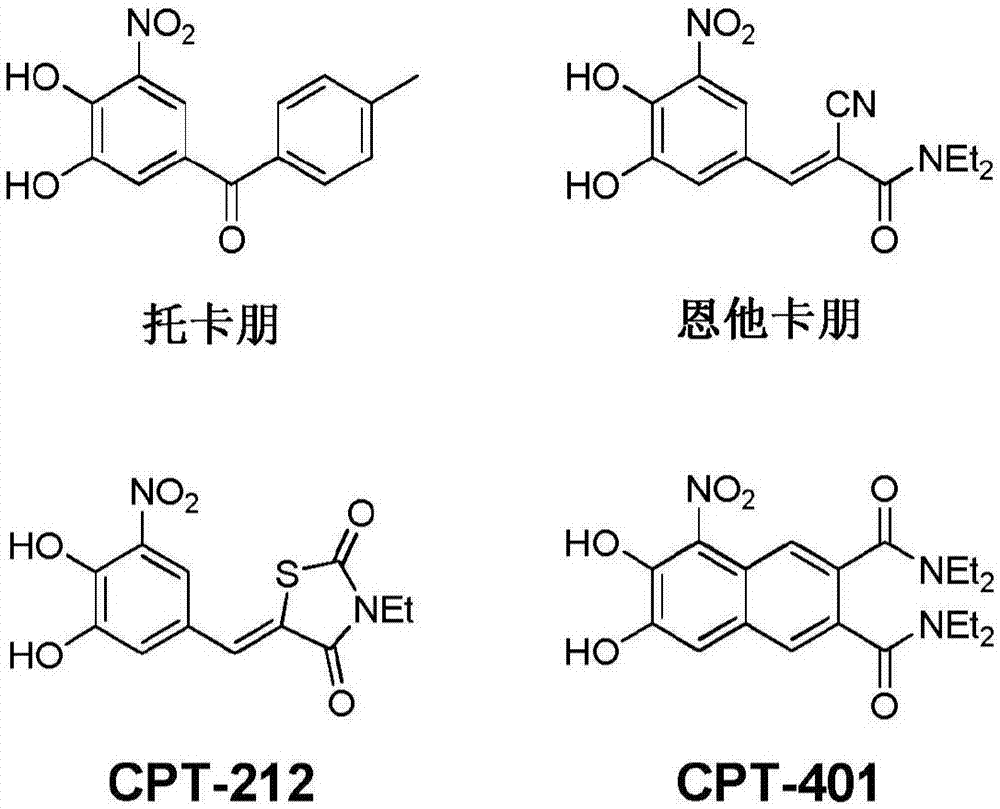
在产生本发明的实验工作中,合成了两种小分子CPT-401(A1)和CPT-212(B1)作为主要COMT抑制剂与托卡朋和恩他卡朋进行比较(图1)。本发明人比较了托卡朋、恩他卡朋和CPT-401(A1)和CPT-212(B1)衍生物对COMT的抑制(图9)。结果显示侧链可以变化而不会损害COMT抑制。
随后,将四种化合物托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)在脱靶结合方面进行比较。
共同治疗靶点的结构相关抑制剂可能对于由不同的脱靶结合引起的潜在毒性问题有所不同。造成托卡朋肝毒性作用的原因可能是其与酶3-羟基异丁酰辅酶A水解酶(HIBCH,CAS NO 9025-88-1)的脱靶结合。HIBCH酶对于通过缬氨酸分解代谢途径中从3-羟基异丁酰辅酶A中去除辅酶A来防止毒性甲基丙烯酰辅酶A的积累是关键的,并且人肝脏中酶活性的降低与肝硬化和肝细胞癌相关,表明处置甲基丙烯酰辅酶A的能力下降可能导致肝衰竭。因此,HIBCH的抑制导致具有细胞毒性的甲基丙烯酰辅酶A的积累。该蛋白质作为托卡朋特异性脱靶的鉴定为肝脏中托卡朋诱导的毒性效应提供了新的可能解释。当与HIBCH结合时,托卡朋采用其中两个芳族部分相互倾斜的构象。相比之下,CPT-401和恩他卡朋表现为相对的平面性。因此,它们与HIBCH的结合被阻止。
儿茶酚O-甲基转移酶(COMT,EC 2.1.1.6)以两种形式存在,即可溶形式(S-COMT)和膜结合形式(MB-COMT)。作为胞质蛋白,S-COMT是大多数组织中的主要形式,并在内源和异生儿茶酚的失活中起更重要的作用。大鼠和人COMT共享81%的序列同一性,并且均属于高度结构保守的SAM依赖性甲基转移酶折叠家族。由于本发明的主要关注点是新型COMT抑制剂在人中的效力,所以在抑制检验中使用内部过表达的重组人COMT蛋白(参见实验程序)。应用先前记载的检验方法(Kurkela,Anal Biochem,2004.331(1):198-200),其中荧光测量在S-(5-腺苷基)-L-甲硫氨酸(AdoMet)存在下在反应混合物中底物七叶素至东莨菪内酯的甲基化。
图2显示化合物CPT-401(A1)、CPT-212(B1)的COMT抑制曲线。在抑制检验中包括托卡朋和恩他卡朋作为参比。据报道,在大鼠肝脏匀浆中,恩他卡朋的IC50为160nM,托卡朋的IC50为36nM(Kiss,J Med Chem,2014.57(21):8692-717)。在使用的检验中,再次发现托卡朋在体外实验条件下比恩他卡朋对于人重组COMT更有效,托卡朋和恩他卡朋的IC50值分别为53nM和386nM。在检验中,领先化合物CPT-401(A1)显示出与托卡朋相似的活性,IC50为54nM,CPT-212(B1)显示IC50为179nM。因此,CPT-401(A1)对COMT的抑制作用与托卡朋相似。CPT-212(B1)表现出比恩他卡朋更高的COMT抑制作用,但显著低于CPT-401(A1)和托卡朋(参见图2)。
副作用通常是由药物与其他蛋白质的脱靶结合引起的。与恩他卡朋相比,托卡朋的严重副作用可能是由于某些蛋白质与托卡朋,而不与恩他卡朋的相互作用。因此,本发明人使用差异竞争捕获化合物质谱(dCCMS)鉴定了与托卡朋相互作用但不与恩他卡朋相互作用的蛋白质。详细信息参阅实验部分。
鉴定出酶3-羟基异丁酰辅酶A水解酶(HIBCH)与托卡朋和CPT-212(B1)结合,但不与恩他卡朋和CPT-401(A1)结合(图5、图6)。这可以通过两个分子对的共同结构特征来解释:托卡朋和CPT-212(B1)采用其中两个芳族部分相互倾斜的构象。该构象整齐地装入HIBCH结合袋(数据未显示)。恩他卡朋、甚至还有CPT-401(A1)优先或限制于整体平面构象。
本发明人比较了托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)的线粒体细胞毒性。已证明托卡朋和CPT-212(B1)的线粒体细胞毒性显著高于恩他卡朋和CPT-401(A1)(图7)。
本发明的化合物、即新型COMT抑制剂CPT-401(A1)与托卡朋相比表现出降低的脱靶结合,并且与恩他卡朋相比表现出增加的效力。因此,它是一种有效的新型COMT抑制剂,具有低线粒体细胞毒性,并且是治疗帕金森氏病的有希望的化合物。
本发明化合物的其他适应症为是阿尔兹海默症、多重耐药性结核病以及肥胖和代谢疾病。
除抑制COMT外,已显示恩他卡朋和托卡朋抑制α-突触核蛋白单体的聚集。这构成了治疗帕金森氏病的第二个有益效果。
COMT抑制剂恩他卡朋和托卡朋还已显示可能通过与原纤维的直接相互作用来抑制Aβ42原细纤维向成熟淀粉样原纤维的聚集(Di Giovanni,J Biol Chem.2010年5月14日;285(20):14941-54)。因此,CPT-401(A1)可用于治疗和预防阿尔兹海默症。
已显示COMT抑制剂托卡朋和恩他卡朋结合并抑制烯酰-酰基载体蛋白还原酶(InhA),即由现有结核药物靶向的酶(Kinnings,PLoS Comput Biol.2009年7月;5(7):e1000423)。InhA对于II型脂肪酸的生物合成和随后的细菌细胞壁的构建必不可少。如异烟肼和乙硫异烟胺等现有的结核病药物结合相同的靶点,但具有完全不同的化学结构并可引起严重的副作用。另外,日益观察到针对这些药物的细菌耐药机制。因此,CPT-401(A1)可用于治疗多重耐药(MDR)和广泛耐药(XDR)结核病。
已显示COMT抑制剂恩他卡朋结合并抑制脂肪量和肥胖相关基因产物(FTO)。恩他卡朋的施用导致甘油三酯合成减少,体重降低和血清胆固醇水平降低(WO2014082544)。因此,CTP-401(A1)可用于肥胖治疗。
实验部分:
合成:
除非另有说明,否则所有反应均在氩气下在干燥的玻璃器皿中进行。市售的高级试剂和溶剂在没有进一步纯化的情况下直接使用。使用预填充的“GraceResolvTM Silica”柱作为固定相,在来自Teledyne Isco的“CombiFlash”Companion系统上进行柱层析。在Buechi系统(Buechi控制单元C-620;两个Buechi泵模块C-605;柱omnifit(230×15mm)上进行MPLC纯化;固定相LiChroprep RP-select B 25–40μm;Buechi UV光度计C-635;Buechi流分收集器C-660;流动相A:0.1%AcOH(Millipore级);流动相B:MeCN(presolv级)。在通过ESI源与Bruker amaZon Speed离子阱耦合的Dionex Ultimate 3000RS HPLC上进行LC-MS。LC方法:将化合物在20%MeCN中稀释至约5μM,将其中50μL用来注入。样品在Phenomenex Luna PFP柱(50×2mm,3μm,)上分离,在5.5分钟内线性梯度为10%至100%B。流动相A:0.1%HCOOH(Millipore级);流动相B:MeCN(LC MS级)。在来自Bruker的Avance 400(400MHz)上记录质子核磁共振(1H NMR)光谱和碳核磁共振(13C NMR)光谱。对于1H-NMR,化学位移相对于d-DMSO(δ2.50)和CDCl3(δ7.27)以ppm报道。
两种新型抑制剂CPT-401(A1)和CPT-212(B1)的合成分别在方案1和2中描述。
化合物CPT-212(B1)可通过哌啶促进的烷基噻唑烷-2,4-二酮衍生物3与市售3,4-二羟基-5-硝基苯甲醛1的缩合反应(US2006142303,通过引用并入本文)获得(方案1)。
方案1.新型COMT抑制剂CPT-212的合成。试剂和条件:(a)乙基溴,DIPEA,DMF,23℃,4天,89%;(b)哌啶,MeOH,80℃,17h,66%.
双环硝基-儿茶酚CPT-401(A1)由市售3,4-二甲氧基苯甲酸4开始,以八个合成步骤合成(方案2)。
通过随后在酸性条件下与甲醛的环化反应和还原性开环将化合物4转化为二醇6(Maziane,Tetrahedron Letters,2001,42(6),1017-1020)。从1,2-双(氯甲基)-4,5-二甲氧基苯开始和随后用乙酸和皂化进行双取代的替代路线似乎更可行,但产率较低。然而,双醇6的Swern氧化以良好的产率得到双醛7,而二氧化锰促进的氧化主要得到内酯5。然后通过四溴化物8并与马来酸酐进行环化反应将双醛7转化成三环化合物9(McOmie,Synthesis,1973,416-417)。用二甲胺使酐9开环以形成单酰胺中间体,并且筛选潜在的偶联试剂表明HATU在两个合成步骤中二酰胺10的产率优异,为62%。三溴化硼促进的双醚切割得到11(Naito,Chemical&Pharmaceutical Bulletin,1991,39(7),1736-1745),其通过选择性引入一个硝基转化为硝基-儿茶酚CPT-401(A1)(Lemaire,Tetrahedron,43(5),835-844)。对于后一种转化来说,硝酸和ACN看起来不能如预期那样工作,并且2,3,5,6-四溴-4-甲基-4-硝基环己-2,5-二烯酮主要产生双硝基副产物。仔细滴定试剂量,最终以适中的收率得到所需产物。CPT-401(A1)的结构可以通过二维NMR光谱和质谱确认。
方案2.新型COMT抑制剂CPT-401(A1)的合成。试剂和条件:(a)甲醛,HCl(气体),70℃,4天,93%;(b)LiAlH4,THF,66℃,4h,77%;(c)草酰氯,DMSO,TEA,DCM,-78℃,105分钟→23℃,60分钟,75%;(d)PBr5,苯,23℃,17h,41%;(e)马来酸酐,NaI,DMF,70℃,48h,69%;(f)二乙胺,HATU,DIPEA,DMF,23℃,48h,62%;(g)BBr3,DCM,0℃,20分钟→23℃,60分钟,98%;(h)2,3,5,6-四溴-4-甲基-4-硝基环己-2,5-二烯酮,乙醚,23℃,60分钟,55%。
化合物3.将噻唑烷-2,4-二酮2(500mg;4.27mmol)和乙基溴(465mg;4.27mmol)在无水DMF(11.4mL)中的溶液用DIPEA(2.23mL;12.81mmol)处理并在23℃下搅拌17小时。由于TLC表明转化不完全,加入另一批乙基溴(465mg;4.27mmol),并将反应混合物在23℃再搅拌3天。减压除去所有挥发物,并通过CombiFlash(环己烷/EtOAc,99/1至3/1)纯化粗产物以获得作为无色液体的化合物3(550mg;3.79mmol;89%)。1H NMR(CDCl3):δ3.94(s,2H),3.69(q,J=7.1Hz,2H),1.20(t,J=7.1Hz,3H)。
化合物CPT-212(B1).将3,4-二羟基-5-硝基苯甲醛1(185mg;1.01mmol)和化合物3(148mg;1.02mmol)在无水MeOH(2.5mL)中的溶液用哌啶(155μL;1.57mmol)处理并在80℃下搅拌17小时。反应混合物用乙酸乙酯稀释并用0.5M HCl溶液洗涤两次。将有机层用Mg2SO4干燥并在减压下除去所有溶剂。粗产物用MeOH重结晶以获得作为黄色固体的硝基-儿茶酚CPT-212(207mg;0.67mmol;66%)。1H NMR(CDCl3):δ10.85(s,1H),7.84(s,1H),7.76(s,1H),7.38(s,1H),6.09(s,1H),3.83(q,J=7.1Hz,2H),1.28(t,J=7.1Hz,3H).LC-MS:rt=3.27分钟;308.83[M-H]-;640.92[2M+Na-2H]-。
化合物5.在0℃下,HCl气体鼓泡通过甲醛溶液(37%的水溶液;90mL;1.2mol)30分钟。加入3,4-二甲氧基苯甲酸4(10g;54.9mmol)并将反应混合物加热至70℃,同时继续使HCl气体鼓泡通过悬浮液。3天后,减压除去所有挥发物并加入水(200mL)。通过添加NH3水溶液将pH调节至pH=7。通过过滤分离固体产物,减压干燥以获得作为灰白色固体的内酯5(9.88g;50.9mmol;93%)。1H NMR(d-DMSO):δ7.26(s,1H),7.23(s,1H),5.27(s,2H),3.87(s,3H),3.84(s,3H)。
化合物6.将内酯5(7.0g;36.0mmol)在无水THF(170mL)中的溶液逐滴添加至氢化铝锂(3.67g;108mmol)的无水THF(50mL)悬浮液中。将反应混合物在66℃下搅拌4小时。冷却至0℃后,通过随后加入水(7mL)和5M NaOH溶液(7mL)小心猝灭反应。过滤后丢弃固体,并在减压下除去所有溶剂。加入水并将产物用DCM充分萃取。合并的有机层用Mg2SO4干燥,减压除去DCM,得到醇6(5.5g;27.7mmol;77%)。1H NMR(d-DMSO):δ6.98(s,2H),4.99(br,s,2H),4.47(s,4H),3.74(s,6H)。
化合物7.在-78℃下,将醇6(2.2g;11.1mmol)在无水DCM(25mL)中的溶液加入预先搅拌的草酰氯(1.94mL;22.2mmol)和DMSO(3.47mL;48.8mmol)在无水DCM(55mL)中的溶液并在-78℃搅拌45分钟。逐滴加入三乙胺(21.84mL;155mmol),并将反应混合物在-78℃再搅拌60分钟,然后升温至23℃并再搅拌60分钟。通过加入水(200mL)小心猝灭反应,并将产物用DCM萃取。将合并的有机层用氯化铵溶液洗涤,用Mg2SO4干燥,并在减压下除去溶剂,得到醛7(1.62g;8.36mmol;75%)。1H NMR(d-DMSO):δ10.52(s,2H),7.54(s,2H),3.94(s,6H)。
化合物8.用五溴代正膦(8.87g;20.6mmol)处理醛7(1.6g;8.24mmol)的无水苯(55mL)溶液并在23℃下搅拌17小时。通过加入1%KOH溶液(50mL)小心猝灭反应。分层,水层用DCM萃取两次。将合并的有机层用盐水洗涤,经Mg2SO4干燥并在减压下除去所有溶剂。通过CombiFlash(环己烷/EtOAc,9/1至1/1)纯化粗产物以获得溴化物8(1.61g;3.34mmol;41%)。1H NMR(CDCl3):δ7.10(br,s,4H),3.93(s,6H)。
化合物9.用碘化钠(2.34g;15.61mmol)处理溴化物8和(1.6g;3.32mmol)和马来酸酐(0.78g;7.97mmol)在无水DMF(20mL)中的溶液,并在70℃搅拌48小时。加入水(100mL)和饱和Na2SO3溶液(20mL)淬灭反应。过滤并减压干燥后获得固体产物9(591mg;2.29mmol;69%)。1H NMR(d-DMSO):δ8.52(s,2H),7.76(s,2H),3.97(s,6H)。
化合物10.用二乙胺(152μL;1.46mmol)处理三环化合物9(171mg;0.662mmol)在无水DMF(200μL)中的溶液并在23℃下搅拌1小时。加入HATU(302mg;0.80mmol)和DIPEA(231μL;1.32mmol)并在23℃下继续搅拌48小时。减压除去所有挥发物,并通过MPLC(水/乙腈,4/1至1/1)纯化粗产物以获得二酰胺10(158mg;0.409mmol;62%)。1H NMR(CDCl3):δ7.59(s,2H),7.10(s,2H),4.01(s,6H),3.52(q,J=7.1Hz,4H),3.31(q,J=7.1Hz,4H),1.24(t,J=7.1Hz,6H),1.10(t,J=7.1Hz,6H)。
化合物11.在0℃下,用BBr3溶液(1M在DCM中的溶液;2.04mL;2.04mmol)处理二酰胺10(158mg;0.409mmol)在无水DCM(16mL)中的溶液。将反应混合物在0℃下搅拌20分钟,使其升温至23℃并再搅拌60分钟。通过氮气流除去所有挥发物,通过MPLC(水/乙腈,4/1至1/9)纯化粗产物以获得二酰胺11(143mg;0.399mmol;98%)。1H NMR(d-DMSO):δ9.73(br,s,2H),7.50(s,2H),7.17(s,2H),3.37(q,J=6.8Hz,4H),3.15(q,J=6.8Hz,4H),1.09(t,J=6.8Hz,6H),1.04(t,J=6.8Hz,6H).1H NMR(CDCl3):δ8.11(br,s,2H),6.83(s,2H),3.54(q,J=6.4Hz,4H),3.27(q,J=6.4Hz,4H),1.25(t,J=6.4Hz,6H),1.07(t,J=6.4Hz,6H)。
化合物CPT-401(A1).随后用2,3,5,6-四溴-4-甲基-4-硝基环己-2,5-二烯酮(9.81mg;0.021mmol)在无水乙醚(1mL)中的溶液将二酰胺11(15.0mg;0.042mmol)在无水乙醚(2mL)中的溶液处理三次,并在23℃下搅拌20分钟。由于TLC显示完全转化,所有挥发物通过氮气流除去并且粗产物通过MPLC(水/乙腈,9/1至3/1)纯化以获得硝基-儿茶酚CPT-401(9.2mg;0.023mmol;55%)。1H NMR(CDCl3):δ12.55(br,s,1H),8.76(s,1H),7.58(s,1H),7.50(s,1H),3.55–3.32(m,4H),3.29(q,J=7.2Hz,4H),1.24(t,J=6.9Hz,6H),1.16(t,J=7.2Hz,3H),1.12(t,J=7.2Hz,3H).LC-MS:rt=2.66分钟;330.98[M-N(CH2CH3)2]+;404.07[M+H]+;807.15[2M+H]+;829.27[2M+Na]+。
通过CCMS鉴定COMT抑制剂的靶向和脱靶蛋白
使用所谓的差异竞争实验分析了调整的实验装置中的托卡朋的特异性结合剂,这些实验跨越了一大组的重复样品、剂量相关性CCMS实验和改进的质谱数据分析。该研究的焦点是来自人肝细胞系HepG2的全细胞裂解物。如前所述,预计儿茶酚部分靶向COMT结合袋,而托卡朋的苄基侧在蛋白质外突出并指向与儿茶酚基团相反的方向。
药物的不同部位可以与诱导不同反应的细胞蛋白产生不同的相互作用。因此,本研究中使用了两个托卡朋选择性功能与捕获化合物支架的连接点(CPT-224:活性Tcp-CC,CPT-220:无活性Tcp-CC,图3)。在活性取向中,捕获化合物应该能够与已知靶标COMT进行特异性相互作用,而在其他取向上,仍然可能与一些脱靶蛋白发生相互作用。
为了测试捕获化合物对COMT的活性,进行了针对内部表达的重组人COMT蛋白的COMT抑制检验。
正如预期的那样,其中托卡朋通过儿茶酚部分连接的具有相反取向的捕获化合物(CPT-220)与COMT没有亲和力。通过托卡朋的苄基部分连接的捕获化合物(CPT-224)使得硝基儿茶酚部分自由地与靶标相互作用,对重组人类COMT显示648nM的IC50,相当于与母体分子相比活性降低约10倍。
通过在肝癌衍生的细胞系HepG2中进行CCMS实验来研究小COMT抑制剂的蛋白质相互作用谱。使用浓度为1μM的无活性捕获化合物(CPT-220)和活性捕获化合物(CPT-224)进行特异性相互作用蛋白的富集和分离。在差异竞争对照实验中,添加过量的各自的游离小COMT抑制剂(托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1))以50μM终浓度与捕获化合物竞争特异性结合剂。使用高分辨率LTQ Orbitrap杂交质谱仪鉴定捕获的并经胰蛋白酶消化的蛋白质。只有鉴定为具有超过99%概率的蛋白质和至少两种具有95%概率的独特肽才被认为鉴定为具有足够的置信度。在竞争对照实验中,加入过量的相应的游离药物,其与相应的CC竞争特定的结合位点。
如预期的那样,使用无活性的捕获化合物(CPT-220)未鉴定出COMT,三种蛋白质符合我们的标准并且被所有四种不同竞争剂竞争,这些蛋白质是吡哆醛激酶(PDXK)、醛-酮还原酶家族1成员B10(AKR1B10)和蛋白质NipSnap同系物3A(NIPSNAP3A)(表1)。当应用活性捕获化合物(CPT-224)时,COMT是最强鉴定的蛋白,以及其他三种蛋白质(犬尿氨酸-氧戊二酸转氨酶3(CCBL2)、1,2-二羟基-3-酮-5-甲硫基戊烯二加氧酶(ADI1)和戊二酰辅酶A脱氢酶(GCDH)一起,表1),其满足发明人的标准并且被所有四种不同的竞争剂竞争。
主要关注的是当使用活性托卡朋捕获化合物时显示出不同竞争特异性的蛋白质,并且这种所谓的差异竞争dCCMS使得能够鉴定该小分子COMT抑制剂的潜在脱靶引起的毒性。在实验中,当本发明人使用四种竞争剂比较特异性鉴定的蛋白质时,蛋白质HIBCH显示独特的捕获模式。当托卡朋被用作竞争剂时,HIBCH被特异性鉴定,而在恩他卡朋存在下没有观察到竞争。有趣的是,当使用CPT-212(B1)作为竞争剂时,HIBCH被特异性竞争,当使用CPT-401(A1)时没有观察到竞争。图5A显示了使用四种不同COMT抑制剂的HIBCH强度的捕获模式。
表1:通过使用活性捕获化合物(上4个小图)和无活性托卡朋捕获化合物(下3个小图)的CCMS实验显著富集并且被全部四种COMT抑制剂竞争的蛋白质。
小分子与HIBCH结合的验证
为了进一步研究托卡朋与HIBCH的结合,进行剂量相关性捕获。图5B中显示了使用5μM至40nM不同浓度的CPT-220或CPT-224和作为竞争剂的托卡朋或CPT-401(A1)的剂量相关性捕获实验的结果。如预期的那样,除了在最高浓度的CPT-220(5μM)下的弱非特异性相互作用之外,COMT蛋白质再次仅以活性取向的捕获化合物捕获,在两个竞争剂(托卡朋和CPT-401(A1))中观察到完全竞争。HIBCH蛋白在不同取向方面观察到相同的现象,在托卡朋-CC(CPT-220)的无活性取向中未鉴定到HIBCH。与此相反,HIBCH只与活性取向的托卡朋-CCP(CPT-224)竞争,但不与CPT-401(A1)竞争。本实验中明确的剂量相关性和结合强度的一致性竞争模式进一步增强了dCCMS检验的有效性。
捕获化合物的独特之处在于捕获化合物与靶蛋白之间形成共价键,并且靶蛋白的结合在小分子存在下发生竞争,所述小分子结合至靶蛋白的相同结合袋。本发明人在HepG2裂解物中进行捕获检验,然后进行抗HIBCH和抗COMT蛋白印迹以通过质谱非相关性读出方法来研究不同竞争物对这些蛋白质的结合谱。图6A显示HIBCH被CPT-224捕获,当托卡朋或CPT-212(B1)用作竞争剂时,捕获被消除,但恩他卡朋或CPT-401(A1)不会。此外,CPT-224与HIBCH的直接交联通过链霉亲和素蛋白印迹(图6B)证实,其在CC交联后检测靶蛋白的生物素化。使用COMT蛋白印迹进行相同的实验(图6C)。仅在使用活性捕获化合物的检验中观察到与COMT的结合,并且当四种小分子托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)中的任一种用作竞争剂时,信号被消除,而且COMT的生物素化在四种竞争剂的存在下被消除(图6D)。该观察结果与质谱数据集(图5A和5B)相关性很好,这证实托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)都是有效的COMT抑制剂,并且CPT-212(B1)与HIBCH的结合可能在与托卡朋结合至HIBCH相同的结合袋中发生。
为了验证这个假设,进行了所有四种小分子与COMT和HIBCH的对接实验。两种新型COMT抑制剂CPT-212(B1)和CPT-401(A1)适合于COMT的结合袋(活性部位)。与此相反,只有弯曲的结构(如托卡朋和CPT-212(B1))可以深“潜”到HIBCH的小分子结合袋中,其中儿茶酚与催化谷氨酰胺酸侧链相互作用,而恩他卡朋和CPT-401(A1)不会,即使对接对于儿茶酚部分的位置受到限制。这可以通过两个分子对的共同结构特征来解释:托卡朋和CPT-212(B1)采用其中两个芳香族部分彼此倾斜,完全装入HIBCH小分子结合袋的构象。但其他两种抑制剂恩他卡朋和CPT-401(A1)优选甚至受限于获得总体平面构象。
COMT抑制剂的细胞毒性结果
在先前的出版物中,本发明人报道了恩他卡朋蛋白结合剂的细胞分布显示与线粒体功能无关,但大部分托卡朋蛋白结合剂是线粒体来源的,特别是线粒体膜(Fischer,Toxicol Sci,2010.113(1):243-53)。为了评估新型COMT抑制剂在基于细胞的检验中可能的线粒体毒性,进行了线粒体ToxGloTM检验,其中HepG2细胞在含半乳糖或含葡萄糖的培养基中生长以基于测量细胞的ATP含量来检测线粒体毒性。ATP产量的减少仅与含半乳糖培养基中的生长有关,因此可能是线粒体功能障碍的结果。除了托卡朋和恩他卡朋之外,在分析中还包括两种新型小分子COMT抑制剂CPT-401(A1)和CPT-212(B1)。如上所述,它们是有效的COMT粘合剂,但在其脱靶特征方面不同。CPT-212(B1)的特征与托卡朋的特征类似,CPT-401(A1)与恩他卡朋的特征类似。
该检验的结果(图7)显示,对于托卡朋来说,在半乳糖培养基中观察到的IC50值比葡萄糖培养基低超过4倍,这表明托卡朋是线粒体毒物。与此相反,观察到恩他卡朋的作用小得多。这些结果进一步支持了由本发明人进行的先前研究的发现,其揭示了托卡朋靶蛋白的线粒体局域化。有趣的是,当比较CPT-401(A1)和CPT-212(B1)的线粒体毒性时,半乳糖和葡萄糖培养基对CPT-401(A1)的IC50值的变化远小于CPT-212(B1),这表明CPT-401(A1)的线粒体毒性小于CPT-212(B1)。
COMT和HIBCH的过表达
His-S-COMT表达载体购自Origene(NM_007310),并且为了克隆HIBCH,使用RNeasy Mini Kit(Qiagen)从HepG2细胞分离RNA,其中使用SuperScript III逆转录酶(Invitrogen)合成cDNA。使用HepG2cDNA作为模板,用以下引物进行PCR反应:5'-gatcgaattcatggggcagcgcgagatg-3'(HIBCH正向),5'-gatcctcgagtcaaaatttcaaatcactgcttcccaaa-3'(HIBCH反向),插入EcoRI和XhoI限制性位点。用下列引物进行G3PDH PCR反应:5'-tgaaggtcggagtcaacggatttggt-3'(G3PDH正向)和5'-catgtgggccatgaggtccaccac-3'(G3PDH反向)作为对照平行进行。使用peqGOLD凝胶提取试剂盒(peqlab)纯化PCR和限制性产物。在FastDigest限制性内切酶和FastDigest Green缓冲液(Thermo Scientific)存在下,在37℃下对PCR片段和靶载体(pET28a+,Merck Millipore)进行2小时限制性酶切。1小时40分钟后,向载体反应物中加入CIP酶,将其在37℃再温育20分钟。使用T4DNA连接酶(Roche)将连接反应在16℃温育过夜。使用具有S-COMT和HIBCH的500ng DNA构建体转化感受态BL21大肠杆菌细胞,并通过IPTG诱导重组蛋白的表达。离心收集细胞(15分钟,4000g,4℃)。将细胞重悬于5ml缓冲液(COMT:50mM磷酸盐,300mM NaCl,5mM MgCl2,pH 7.4,HIBCH:20mM Tris,500mM NaCl,10mM咪唑,0.5%(w/v)Triton X-100,pH 8.0)并通过用溶菌酶和DNA酶I处理然后超声破碎。来自随后离心的上清液(细胞溶质部分)在随后的实验中用作蛋白质来源。
COMT活性微板检验
COMT活性测量实验基本上如先前所述进行(Kurkela,Anal Biochem,2004.331(1):198-200)。简单来说,将七叶素(Sigma-Aldrich)和竞争性儿茶酚底物溶解于二甲基亚砜(DMSO)中并用缓冲水溶液(100mM磷酸盐,5mM MgCl2,20mM L-半胱氨酸,pH 7.4)稀释以获得最终的在100μl反应混合物中为2%的DMSO浓度。所有的试剂溶解在相同的缓冲溶液中。对于每个样品,将抑制剂和七叶素溶液一式三份移液到黑色圆底96孔板中。在每个微孔板中包含不含抑制剂或AdoMet的对照。将板置于冰上,加入含有酶的细胞溶质部分,至15μg/ml的最终蛋白质浓度。通过将微板置于37℃开始5分钟的预温育时间,通过在37℃下加入AdoMet开始反应,最终浓度为10μM。在反应开始时和在60分钟时使用CARY Eclipse荧光分光光度计测量荧光。激发和发射波长分别为355nm和460nm。通过从60分钟时的荧光减去检验开始时的荧光来估算酶活性,并使用GraphPad Prism产生抑制曲线。
捕获实验
捕获实验用500μg的HepG2裂解物(InVivo,Germany)或1μg的人重组COMT或1μg人重组HIBCH蛋白进行,其补充有5μM AdoMet(Sigma-Aldrich)、20μl的5×捕获缓冲液(caprotec bioanalytics GmbH),并与DMSO或50μM竞争剂在4℃的100μl总体积中温育30分钟。在4℃与捕获化合物温育1小时后,使用caproBox(Caprotec Bioanalytics GmbH)照射样品10分钟。随后,通过向每个反应添加25μL的5×洗涤缓冲液(WB,caprotec bioanalytics GmbH,Berlin,Germany)来调节缓冲液条件。然后将样品与50μL Dynabeads MyOne链霉亲和素C1(Invitrogen,Karlsruhe,Germany)在4℃下在旋转轮上温育1小时。当使用磁珠(caprotec bioanalytics GmbH,Berlin,Germany)时,然后使用caproMag(用于方便且有效地分离生物分子的设备)收集含有CC-蛋白质缀合物的珠子,并且用200μL的1×WB冲洗六次,然后用MS级水冲洗。将珠子在4℃下保存在去离子水中,直至通过SDS-PAGE或蛋白质消化然后通过LC-MS进行进一步分析。
蛋白质消化和质谱分析
蛋白质捕获后,链霉亲和素磁珠用200μL的LC-MS级水(Fluka,St.Louis,MO,USA)洗涤两次。对于胰蛋白酶消化,将结合至链霉亲和素磁珠的蛋白质与9μL的50mM碳酸氢铵和1μL胰蛋白酶(0.5μg/μL)(Roche,Germany)一起在温度控制振动筛上在37℃温育16小时。取出上清液,在miVac DNA真空离心机(Genevac,UK)中蒸发至干并直接用于质谱分析。胰蛋白酶消化物在通过Proxeon纳米电喷雾离子源(Thermo Fisher Scientific,Germany)与LTQ-Orbitrap Velos仪器(Thermo Fisher Scientific,Germany)连接的UltiMate 3000RSLCnano系统上通过在线纳升液相色谱串联质谱法进行分析(nl LC-MS/MS)。对于色谱分离,首先将样品加载到反相(RP)预柱(Acclaim PepMap100)上并在RP分析柱(Acclaim PepMap RSLC C18,Dionex,Thermo Fisher Scientific,Germany)上分离。质谱分析以数据相关模式进行,允许Orbitrap-MS和LTQ-MS/MS采集之间的自动切换,以进行全扫描和后续的碰撞诱导解离(CID)碎裂。在Orbitrap分析仪中采集全扫描质谱(m/z 300-2000)。使用CID在线性离子阱中依次分离和碎裂20个最强烈的离子。
使用MaxQuant(www.maxquant.org,版本1.4.0.8)中实施的搜索引擎Andromeda分析所有MS/MS数据。使用6ppm前体公差,0.5Da片段离子公差,完全胰蛋白酶特异性允许多达2次漏失的切割和甲硫氨酸氧化作为可变修改进行针对人类UniProtKB/Swiss-Prot数据库的自动数据库搜索(版本2015_02包含20203被复查的序列条目)。在蛋白质和肽水平上,最大错误发现率均设为0.01,并且要求6个氨基酸作为最小肽长度。选择无标记定量选项用于LC-MS/MS运行之间的比对。根据在caprotec bioanalytics GmbH实施的标准化程序对MaxQuant分析的输出进行后处理,以使蛋白质强度归一化,并随后计算倍数变化和p值。
因此,在捕获检验中通过LC-MS检测到并且在竞争对照实验中显著减少的蛋白质被定义为特异性的。通过每种蛋白质的检验和竞争之间的倍数变化来量化特异性。此外,实验一式四份进行,允许通过计算p值来确定显著性。
ATPlite细胞毒性检验
由Pharmacelcus GmbH进行化合物托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)的线粒体毒性评估(Glu/Gal)。简言之,将在葡萄糖培养基(含有22mM葡萄糖、1mM丙酮酸钠、5mM Hepes、2mM L-谷氨酰胺、10%FCS和1%青霉素/链霉素的高葡萄糖DMEM)或半乳糖培养基(含有10mM半乳糖、1mM丙酮酸钠、5mM Hepes、2mM L-谷氨酰胺、10%FCS和1%青霉素/链霉素的高葡萄糖DMEM)中培养的HepG2细胞加入到96孔板(100μL,4×104个细胞/孔)中。对于药物治疗,在DMSO中制备化合物储备溶液并将其加入孔中以得到所述的最终药物浓度。加入发光ATP检测试剂后各孔中DMSO的最终浓度为0.5%。每孔加入总共100μL的发光ATPlite(PerkinElmer)检测试剂。在平板振荡器上短暂混合后,将反应在室温下温育2分钟,使用Victor X5平板读数器在2分钟后测量发光。
TMRE线粒体膜电位检验
对于TMRE线粒体膜电位检验,用不同浓度的四种COMT抑制剂处理HepG2细胞。TMRE(四甲基罗丹明乙酯)是一种细胞可渗透的带正电的红橙色染料,由于线粒体相对较负的电荷而容易在活性线粒体中积累,该染料用于标记活性线粒体。观察到四种COMT抑制剂之间线粒体膜电位状态的显著差异(图9)。在50μM的浓度下,化合物托卡朋和CPT212将膜电位抑制至低于未处理的HepG2细胞的20%~30%,而使用化合物恩他卡朋或CPT401仍然观察到超过50%的膜电位。浓度为100μM的CPT212和托卡朋将膜电位降低至对照的0~5%,而对于CPT401和恩他卡朋,在该浓度仍保持40%~50%的膜电位。数据表明恩他卡朋与CPT401以及托卡朋与CPT212在10μM至100μM浓度之间在线粒体膜电位上非常相似。使用带正电的荧光染料TMRE测量线粒体膜电位(MMP),其容易积聚在活性线粒体中。FCCP是氧化磷酸化的离子载体解偶联剂,用作阳性对照。将HepG2细胞接种到μClear平底黑色96孔板(约4×104个细胞/孔)中。达到汇合后,将细胞在37℃和5%CO2下用单独确定的最终浓度的四种COMT抑制剂(托卡朋、恩他卡朋,CPT401或3)处理过夜。将培养基移出并用新鲜培养基替换,将TMRE以200nM终浓度添加至培养基,并将细胞在37℃和5%CO2下温育30分钟。温育后,用含有0.2%BSA的PBS洗涤细胞,使用分别配备有549nm和575nm激发和发射滤光片的Cary Eclipse荧光酶标仪测量荧光强度。
计算方法
所有使用的软件工具都包含在SYBYLx1.2软件包(Tripos Inc.,St.Louis,MO)中。为了准备分子结构,从PDB文件3BUT获得与槲皮素(QUE)和(2R)-3-羟基-2-甲基丙酸(HIU)复合的人β-羟基异丁酰辅酶A水解酶(HIBCH_HUMAN)的原子坐标。使用Prepare Protein工具对结构进行预处理,并使用MMFF力场、MMFF电荷和Powell梯度算法(rmsd 0.5)将其最小化。所有重原子在最小化期间都被冻结。去除QUE以访问催化位点。使用配体工具从二维草图(ChemDraw)获得配体结构:结构被标准化,并且生成互变异构体(带电原子的计数=4)。用子结构SMARTS过滤器(O[-1]C:CO[-1])去除儿茶酚二价阴离子,并且使用Concord Standalone生成每种互变异构体的一个3D结构。儿茶酚和硝基的原子类型设置如下:O-->O.co2;N->N.pl3,使用SPL脚本。使用默认设置(Tripos FF,rmsd=0.05)和Gasteiger-Marsili费用使得到的结构最小化。使用Surflex-Dock进行HIBCH与推定的抑制剂托卡朋、恩他卡朋,CPT-212(B1)和CPT-401(A1)之间的无约束灵活对接。提取共结晶产物HIU并基于HIU产生原生质体。允许蛋白质的灵活性(氢原子和重原子)并产生10个启动构象。否则,使用默认设置。对100个姿势进行取样,调查对接评分、儿茶酚位置和蛋白质。托卡朋的最佳对接姿势被用作儿茶酚片段的基础。使用弱限制片段放置(cpen=10)重复对接。
缩写:
CAN,硝酸铈铵;CC,捕获化合物;CCMS,捕获化合物质谱法;dCCMS,差异竞争捕获化合物质谱法;DCM,二氯甲烷;DIPEA,N,N-二异丙基乙胺;DMF,N,N-二甲基甲酰胺;DMSO,二甲基亚砜;HATU,O-(7-氮杂苯并三唑-1-基)-N,N,N’-四甲基脲六氟磷酸盐;TEA,三乙胺;THF,四氢呋喃。
附图说明
图1显示托卡朋、恩他卡朋和两种领先COMT抑制剂CPT-401(A1)和CPT-212(B1)的化学结构。
图2显示托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)对人重组COMT的抑制作用。
图3显示无活性(通过儿茶酚官能连接,CPT-220)和活性(通过具有游离儿茶酚功能的苄基连接,CPT-224)取向的托卡朋捕获化合物的化学结构。
图4显示通过活性托卡朋捕获化合物(CPT-224)和无活性托卡朋捕获化合物(CPT-220)对人重组COMT的抑制。
图5A显示差异竞争捕获化合物质谱实验的结果,其包含在dCCMS实验中鉴定的HIBCH的平均MS/MS和归一化强度:A:检验;C1:托卡朋作为竞争剂;C2:恩他卡朋作为竞争剂;C3:CPT-401(A1)作为竞争剂;C4:CPT-212(B1)作为竞争剂。数据为平均值+SD(n=4)。该表给出了使用CPT-224与不同竞争剂C1:托卡朋,C2:恩他卡朋,C3:CPT-401(A1),C4:CPT-212(B1))的捕获实验中HIBCH蛋白的FC和p值。
图5B显示使用5μM至40nM不同浓度的CPT-220或CPT-224的剂量相关性捕获实验。左图:COMT蛋白的CCMS结果;右图:HIBCH蛋白的CCMS结果;检验:用CPT-220或CPT-224的检验;C1/Tcp:在CPT-220或CPT-224的存在下,托卡朋作为竞争剂;C3/CPT-401:CPT-401(A1)作为竞争剂。数据为平均值+SD(n=4)。
图6显示差异竞争捕获化合物交联后通过蛋白印迹检测COMT和HIBCH:Tcp-CC(CPT-224)捕获重组HIBCH(左图)和重组COMT(右图)(A),抗HIBCH蛋白印迹证明了HIBCH蛋白的鉴定。(B),抗链霉亲和素蛋白蛋白印迹证明了HIBCH与CPT-224的交联。反应在Tcp和CPT-212(B1)存在下竞争,而在Ecp和CPT-401(A1)存在下没有观察到HIBCH竞争。(C),抗COMT蛋白印迹证明了COMT蛋白质的鉴定。D),抗链霉亲和素蛋白印迹证明了COMT与CPT-224的交联。反应在Tcp、Ecp、CPT-401(A1)和CPT-212(B1)的存在下竞争。
图7显示使用托卡朋、恩他卡朋、CPT-401(A1)和CPT-212(B1)的基于细胞的线粒体毒性检验的结果。与含半乳糖培养基中生长有关的ATP输出量的减少表明线粒体功能障碍。X轴:ATP浓度;Y轴:测试化合物的浓度。
图8显示COMT抑制剂的化学结构以及μM和0.1μM浓度的COMT抑制检验的结果。A:托卡朋、恩他卡朋。B:CPT-401(A1)的衍生物。C:CPT-212(B1)的衍生物。
图9显示使用TMRE线粒体膜电位测试托卡朋、恩他卡朋、CPT-212(B1)或CPT-401(A1)对线粒体膜电位(对照百分比)在HepG2细胞中的剂量相关作用。数据是平均SD(n=6)。
- 还没有人留言评论。精彩留言会获得点赞!