茂金属催化剂组合物和使用其的聚合方法与流程
相关申请和专利
本发明涉及:同时提交的pct申请_______(代理人案卷号2016em109),标题为“supportedmetallocenecatalystsystemsforpolymerization”;pct申请_______(代理人案卷号2016em113),标题为“singlereactorproductionofpolymersingasorslurryphase”和pct申请_______(代理人案卷号2016em114),标题为“productionofheterophasicpolymersingasorslurryphase”。
发明领域
本发明涉及新的催化剂化合物,包含非对称取代的茚基的催化剂体系及其用途。
发明背景
烯烃聚合催化剂在工业上有很大的应用。因此,存在着寻找新催化剂体系的兴趣,其增加了催化剂的商业有用性和允许生产具有改进的性能的聚合物。
用于烯烃聚合的催化剂经常基于茂金属作为催化剂前体,其通常是用铝氧烷或者用含有非配位阴离子的活化剂来活化的。然而,用于丙烯共聚物的茂金属催化剂已经受限于它们不能生产高分子量或者其他期望性能的丙烯-乙烯共聚物。许多茂金属结构已经观察到这种现象,例如用于弹性聚丙烯的间规cs对称me2c(cp)(flu)zrcl2,非等规(aspecific)c2v对称me2si(flu)2zrcl2以及c2对称外消旋-me2c(3-ipr-ind)2zrcl2和流动(fluxional)(2-ph-ind)2zrcl2催化剂二者。在等规c2对称外消旋-me2si(2-me-4,5-benz-ind)2zrcl2和外消旋-me2si(2-me-4-ph-ind)2zrcl2中也发现这种不足(l.resconi,c.fritze,“metallocenecatalystsforpropylenepolymerization”,polypropylenehandbook(n.pasquini编辑),第2.2章,出版商hanser,munic2005)。据认为,虽然这种催化剂族的2-me取代抑制了到丙烯单体的β-氢转移和因此防止形成低分子量聚合物,但是它未能防止在后者存在的情况下β-氢转移到乙烯共聚单体。到乙烯共聚单体的这种β-氢转移变成有利的链终止机理和导致形成低分子量丙烯-乙烯共聚物(a.tynys等人,macromol.chem.phys.2005,第206卷,第1043-1056页:“ethylene-propylenecopolymerizations:effectofmetallocenestructureonterminationreactionsandpolymermicrostructure”)。已经在具有大体积配体的一些二茂锆中发现例外,例如外消旋-me2c(3-tbu-ind)2zrcl2,其表现出通过乙烯引入带来的分子量的显著增加。然而,这种催化剂在均聚物分子量和活性方面具有不足。
用于生产全同立构聚丙烯的令人期望的茂金属催化剂生产了具有高熔点的聚丙烯。这被认为归因于聚合物微结构中的高立体定向性和/或区域选择性。在外消旋-alk2si(2-alk-ind)2zrcl2催化剂族(alk=烷基)内,立体定向性和区域选择性是持续改变的。例如,ep834519a1涉及外消旋-me2si(2-me-4-ar-ind)2zrcl2类型茂金属,用于生产硬质高熔点聚丙烯,其具有高的立构规整度和非常低的区域误差量。然而,这些聚丙烯在商业相关方法条件下不是很成功,并且遭受了低活性/生产率水平。
us-a12001/0053833公开了茂金属,其中2-位置是用未取代的杂芳族环或者具有键合到该环上的至少一个取代基的杂芳族环来取代的,其产生了具有小于期望的熔点的丙烯乙烯共聚物。
wo01/058970涉及具有高熔点和良好的橡胶含量的冲击共聚物,其当两个烷基取代基是异丙基时是通过外消旋-me2si(2-alk-4-ar-ind)2zrcl2族催化剂来产生的。然而,这些催化剂遭受了活性问题。
wo02/002576公开了(2-烷基-4-ph-ind)2zrcl2族,其中该2-位置可以是异丙基,和ph取代基是在3和5位置取代的,特别是用叔丁基取代的。然而,这些催化剂在商业条件也遭受了活性/生产率问题。
wo03/002583公开了(2-烷基-4-ph-ind)2zrcl2族,其中2-位置可以用异丙基取代和4位置是用在2-位置取代的ph基团取代的,特别是用苯基取代的。然而,这些催化剂在商业条件也遭受了活性/生产率问题。另外,这些催化剂对于全同立构均聚丙烯具有相对低的mw能力。
ep-a21250365;wo97/40075;和wo03/045551涉及联茚基茂金属,其中在任一茚基配体的2-位置上的取代基是在α-位置支化或者环化的。然而,这些催化剂对于全同立构均聚丙烯仍然具有相对有限的mw能力。
wo04/106351涉及联茚基茂金属,其在茚基配体的2-位置具有取代基,并且条件是一个配体是未支化的或者经由sp2-杂化碳原子键合的,和另一配体是在α-位置支化的。然而,这些催化剂对于全同立构均聚丙烯仍然具有相对有限的mw能力。
us8507706公开了联茚基茂金属,其中茚基上的至少一个2位置是用在β位置支化的基团取代的,和另一2-位置在α位置是未支化的。us2011/0230630公开了类似的茂金属,除了在2位置的基团在β位置是支化的,并且该β-碳原子是季碳原子和是非环状烃体系的一部分。
us7829495公开了烷基取代的茂金属,其具有“通过伯碳原子…键合到la或者lb环任一上的…c3或者更大烃基…取代基…优选正烷基取代基”(参见第4栏第9-12行)。此外,在实施例部分中,(正丙基环戊二烯基)(四甲基环戊二烯基)二氯化锆与甲基铝氧烷和davisiontm948二氧化硅组合用于乙烯己烯聚合;双(正丙基环戊二烯基)二氯化锆与甲基铝氧烷和davisiontm948二氧化硅组合用于乙烯己烯聚合;和二甲基甲硅烷基(芴基)(正丙基环戊二烯基)二氯化锆与甲基铝氧烷和davision二氧化硅组合用于乙烯己烯聚合。
2015年1月22日公开的us2015/0025208公开了桥连联茚基化合物,其中茚上的2位置(r2和r8)是不相同的,和该茚上的4位置(r4和r10)是取代的苯基,其中r4和r10中的至少一个是在3和5位置上取代的苯基。
us2005/0182266公开了制备具有特定取代方式的过渡金属化合物的方法,相应的过渡金属化合物本身和它们在制备催化剂体系中的用途以及该催化剂体系在烯烃的聚合和共聚中的用途。
感兴趣的其他参考文献包括us6051727;us6255506;ep0576970;us5459117;us5532396;us5543373;us5585509;us5631202;us5696045;us5700886;us6492465;us6150481;us5770753;us5786432;us5840644;us6242544;us5869584;us6399533;us6444833;us6559252;us6608224;us6635779;us6841501;us6878786;us6949614;us6953829;us7034173;us7141527;us7314903;us7342078;us7405261;us7452949;us7569651;us7615597;us7799880;us7964679;us7985799;us8222356;us5278264;us5276208;us5049535;us2011/0230630;w002/002575;wo02/022575;wo2003/002583;us7122498;us2011/0230630;us2010/0267907;ep1250365;wo97/9740075;wo03/045551;wo02/002576;us2015/0025205;ussn14/572195;2014年12月16日提交;us9193856;wo2004/052945;us2016/0032025;和journalofmolecularcatalysisa:chemical(20010705),172(1-2),第43-65页。
本发明涉及共同拥有的专利us9249239和共同待决申请ussn15/000731,2016年1月19日提交;ussn14/324333,2014年7月7日提交;ussn14/324408,2014年7月7日提交;和ussn14/324427,2014年7月7日提交。
本领域仍然需要用于烯烃聚合的新的和改进的催化剂体系,来实现特定的聚合物性能,例如高熔点,高分子量,来增加转化率或者共聚单体的引入,或者来改变共聚单体分布,而不劣化所形成的聚合物的性能。
所以本发明的一个目标是提供新的催化剂化合物,包含这样的化合物的催化剂体系,和使用这样的化合物和体系来聚合烯烃的方法。
此外,本发明的目标是提供烯烃聚合物,特别是丙烯均聚物,和丙烯与乙烯和/或高级α-烯烃的无规共聚物。
发明概述
本发明涉及下式所示的茂金属催化剂化合物:
其中,
r2和r8独立地是c1-c20线性烷基,条件是r2和r8中的至少一个必须具有至少4个碳原子;
r4和r10是取代或者未取代的芳基,条件是芳基中的至少一个是:1)在邻位用选自c1-c40烃基、杂原子和含杂原子的基团中的至少一个基团取代的,和/或2)在3'、4'或者5'位置上用选自c1-c40烃基、杂原子和含杂原子的基团的至少一个基团取代的;
m是第2、3或者4族过渡金属;
t是桥连基团;
每个x是阴离子离去基团;
每个r1、r3、r5、r6、r7、r9、r11、r12、r13和r14独立地是氢、或者烃基、取代的烃基、卤代碳基、取代的卤代碳基、甲硅烷基碳基、取代的甲硅烷基碳基、甲锗烷基碳基或者取代的甲锗烷基碳基取代基。
本发明进一步涉及催化剂体系,其包含这样的茂金属和活化剂。
本发明进一步涉及聚合烯烃的方法,其包括将烯烃与包含所述的上述茂金属催化剂化合物(一种或多种)和活化剂的催化剂体系接触。
本发明进一步涉及聚合烯烃的方法,其包括在60℃(供选择地80℃)或者更高的温度,将烯烃与包含活化剂和一种或多种上述催化剂化合物的催化剂体系接触,和优选获得聚合物,其具有:a)0.5-60重量%乙烯,基于共聚物的重量;b)200000g/mol或者更大的mw,其是通过gpc-dri测定的。
附图简要描述
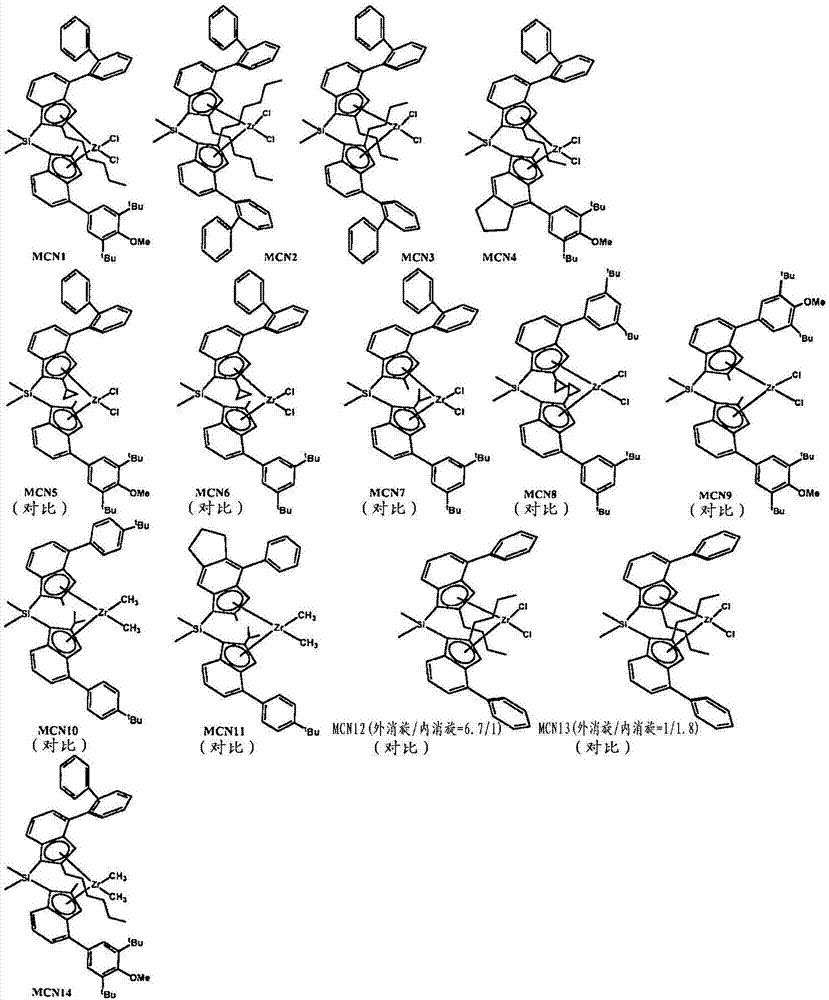
图1是mcn1-14的式的图。
图2是来自于表1的mw对乙烯供料压力的图(数据在两个运行上平均,除了条件“mnc7,120psic2”)。
图3是来自于表2的mw对c2wt%引入率的图。
图4是来自于表3的mw对c2wt%引入率的图。
图5是来自于表4的ipp熔点对聚合温度数据的图(数据在两个运行上平均)。
图6是来自于表4的mw对聚合温度数据的图(数据在两个运行上平均)。
图7是来自于表4的活性对乙烯供料压力数据的图(数据在两个运行上平均)。
图8是来自于表4的mw对乙烯供料压力数据的图(数据在两个运行上平均)。
图9是实施例163(左)和164(右)种所制造的聚合物的gpc-3d图。
图10是实施例163(左)和164(右)种所制造的聚合物的流变测量的图。
图11是实施例163和164中所制造的聚合物的相位角对复数模量的图。
图12是实施例163和164所制造的聚合物的复数剪切粘度对频率的图。
图13是用于合成茂金属的反应方案。
发明详述
就本发明及其权利要求书的目的而言,使用了周期表族的新编号方案,如chemicalandengineeringnews,63(5),第27页(1985)所述。所以,“第4族金属”是周期表第4族的元素。
除非另有指示,否则“催化剂生产率”是使用包含wg的催化剂(cat)的聚合物催化剂,在t小时的时间期间生产了多少克聚合物(p)的度量;并且可以用下式来表示:p/(txw),并且以单位gpgcat-1hr-1来表示。除非另有指示,否则“催化剂活性”是催化剂有多活泼的度量,并且是作为产物聚合物的质量(p)/所用催化剂(cat)的摩尔数(kgp/molcat)来报告的。除非另有指示,否则“转化率”是转化成聚合物产物的单体的量,并且是作为mol%来报告的,并且是基于聚合物收率和供入反应器中的单体的量来计算的。
“烯烃(olefin)”,供选择地称为“烯烃(alkene)”,是具有至少一个双键的线性、支化的或者环状的碳和氢的化合物。就本说明书及其所附的权利要求书的目的而言,当聚合物或者共聚物被称作包含烯烃时,在这样的聚合物或者共聚物中存在的烯烃是烯烃的聚合形式。例如,当共聚物据称“乙烯”含量是35wt%-55wt%时,应当理解该共聚物中的单体单元衍生自聚合反应中的乙烯,并且所述的衍生单元是以35wt%-55wt%存在的,基于共聚物的重量。“聚合物”具有两个或者更多个相同或者不同的单体单元。“均聚物”是具有相同单体单元的聚合物。“共聚物”是具有两种或者更多种彼此不同的单体单元的聚合物。“三元共聚物”是具有三种彼此不同的单体单元的聚合物。用于提及单体单元的“不同的”表示该单体单元彼此相差至少一个原子或者是同分异构不同的。因此,如本文所用的,共聚物的定义包括三元共聚物等。“乙烯聚合物”或者“乙烯共聚物”是包含至少50mol%的乙烯衍生单元的聚合物或共聚物,“丙烯聚合物”或者“丙烯共聚物”是包含至少50mol%的丙烯衍生单元的聚合物或共聚物等。
就本发明的目的而言,乙烯应当被认为是α-烯烃。
就本发明及其权利要求书的目的而言,术语“取代的”表示氢基团已经用杂原子或者含有杂原子基团替代。例如,“取代的烃基”是由碳和氢组成的基团,其中至少一个氢是用杂原子或者含杂原子的基团替代的。
除非另有指示,否则室温是23℃。
用于提及本文中任何式中的r基团(例如r2和r8或者r4和r10)或者本文中任何取代基的“不同的”或者“不相同”表示该基团或取代基彼此相差至少一个原子或者是同分异构不同的。
如本文所用的,mn是数均分子量,mw是重均分子量,和mz是z均分子量,wt%是重量百分比,和mol%是摩尔百分比。分子量分布(mwd),也称作多分散性指数(pdi),定义为mw除以mn。除非另有规定,否则全部分子量单位(例如mw,mn,mz)是以g/mol报告的。下面的缩写可以用于本文:me是甲基,et是乙基,pr是丙基,cpr是环丙基,npr是正丙基,ipr是异丙基,bu是丁基,nbu是正丁基,ibu是异丁基,sbu是仲丁基,tbu是叔丁基,oct是辛基,ph是苯基,bn是苄基,mao是甲基铝氧烷。
“催化剂体系”是至少一种催化剂化合物、至少一种活化剂、任选的助活化剂和任选的载体材料的组合。就本发明及其权利要求书的目的而言,当催化剂体系被描述为包含组分的中性稳定形式时,本领域技术人员公知的是该组分的离子形式是与单体反应来产生聚合物的形式。
在本文的描述中,茂金属催化剂可以描述为催化剂前体,预催化剂化合物,茂金属催化剂化合物或者过渡金属化合物,并且这些术语是可互换使用的。“阴离子配体”是带负电的配体,其将一对或多对电子贡献给金属离子。
茂金属催化剂定义为有机金属化合物,其具有至少一个π-键合的环戊二烯基部分(或者取代的环戊二烯基部分)和更经常地两个π-键合的环戊二烯基部分或者取代的环戊二烯基部分,其键合到过渡金属上。
就涉及茂金属催化剂化合物的本发明及其权利要求书的目的而言,术语“取代的”表示氢基团已经用烃基,杂原子或者含杂原子的基团替代。例如,甲基环戊二烯(cp)是甲基取代的cp基团。
就本发明及其权利要求书的目的而言,“烃氧基(alkoxide)”包括那些,其中烃基(alkyl)是c1-c10烃基(hydrocarbyl)。该烃基可以是直链、支化的或者环状的。该烃基可以是饱和的或者不饱和的。在一些实施方案中,该烃基可以包含至少一个芳基。
作为与本发明的茚基化合物相关使用的“非对称”表示在4位置上的取代是不同的,或者在2位置上的取代是不同的,或者在4位置上的取代是不同的和在2位置上的取代是不同的。
茂金属催化剂化合物
本发明涉及下式所示的茂金属催化剂化合物:
其中,
r2和r8可以是相同或者不同的,并且每个独立地是c1-c20线性烷基,条件是r2和r8中的至少一个具有至少4个碳原子,优选至少6个碳原子,优选r2和r8在α或者β位置不具有支链;
r4和r10是取代或者未取代的芳基(例如取代或者未取代的苯基,优选取代的苯基),条件是芳基中的至少一个是:1)在邻位用选自c1-c40烃基、杂原子和含杂原子的基团中的至少一个基团取代的,和/或2)在3'、4'或者5'位置上用选自c1-c40烃基、杂原子和含杂原子的基团的至少一个基团取代的;
m是第2、3或者4族过渡金属,优选第4族过渡金属;
t是桥连基团;
每个x是阴离子离去基团;
每个r1、r3、r5、r6、r7、r9、r11、r12、r13和r14独立地是氢、或者烃基、取代的烃基、卤代碳基、取代的卤代碳基、甲硅烷基碳基、取代的甲硅烷基碳基、甲锗烷基碳基或者取代的甲锗烷基碳基取代基。
在本文所述的任何实施方案中,r2可以是线性c1-c10烷基,例如甲基,乙基,正丙基,正丁基,正戊基,正己基,正庚基,正辛基,正壬基或者正癸基),其可以是卤代的,优选用i,f,cl或者br卤代。
在本文所述的任何实施方案中,r8是线性c1-c10烷基,优选甲基,乙基,正丙基,正丁基,正戊基,正己基,正庚基,正辛基,正壬基或者正癸基),其可以是卤代的,优选用i,f,cl或者br卤代。
在本发明的一些实施方案中,r2和r8是相同的线性烷基,例如正丁基,正己基等。
在供选择地实施方案中,r2和r8是不同的,例如r2是甲基和r8是正丁基,正戊基,正己基,正庚基或者正辛基。
用“取代的苯基”表示苯基是用1,2,3,4或者5个c1-c20取代或者未取代的烃基取代的,例如甲基,乙基,丙基,丁基,戊基,己基,庚基,辛基,壬基,癸基,十一烷基,十二烷基,苯基,取代的苯基,联苯基或者其异构体。在有用的实施方案中,该苯基是在间或者对位上,优选在3'和/或5'位置上取代的,优选用c4-c12烷基取代。供选择地,该苯基可以在2'位置取代,但是优选在2'和6'位置不取代,例如在本发明的优选的实施方案中,当苯基的2'位置是取代的时,6'位置是h)。供选择地,该苯基可以是在4'位置上用式(xr'n)-的基团取代的,其中x是第14,15,16或者17族杂原子和r'是氢原子,卤素原子,c1-c10烷基或者c6-c10芳基之一,和n是0,1,2或者3;优选–nr'2,-sr',-or',-osir'3,-sir'3或者-pr'2;和任选地,该苯基上的一个或多个其余位置是取代的,例如2',3'和或5'位置。
在另一方面,芳基上的4'位置不是c4基团,供选择地不是烃基。
在另一方面,r4和r10独立地是取代的苯基,优选用下面的基团取代的苯基:c1-c10烷基(例如叔丁基,仲丁基,正丁基,异丙基,正丙基,环丙基,环丁基,环戊基,环己基,环庚基,环辛基,苯基,三甲基苯基,或者金刚烷基),或者芳基,其可以进一步用芳基取代,并且键合在一起的两个芳基可以直接或者通过连接基团结合在一起,其中该连接基团是烷基,乙烯基,苯基,炔基,甲硅烷基,甲锗烷基,胺,铵,膦,鏻,醚,硫醚,硼烷,硼酸根,铝烷(alane)或者铝酸根基团。
在另一方面,r4和r10的至少一个(或者任选地,二者)是在3'和5'位置取代的苯基。
在另一方面,r4和r10的至少一个(或者任选地,二者)是在2'位置用烷基或者芳基例如苯基取代的苯基。
在另一方面,r4和r10的至少一个(或者任选地,二者)是在3'和5'位置取代的苯基,和r4和r10的至少一个是在2'位置用烷基或者芳基例如苯基取代的苯基。
在再一方面,r4和r10的至少一个(或者任选地,二者)是在3'和5'位置用c1-c10烷基例如叔丁基取代的苯基。
在再一方面,r4和r10的至少一个是在3'和5'位置用c1-c10烷基例如叔丁基取代的苯基,和r4和r10的至少一个是在2'位置用烷基或者芳基例如苯基取代的苯基。
在再一方面,r4和r10的至少一个是在3'和5'位置用c1-c10烷基例如叔丁基取代,和在4'位置用(xr’n)-取代的苯基,其中x是第14,15,16或者17族杂原子,其原子量是13-79,r’是氢原子,卤素原子,c1-c10烷基或者c6-c10芳基之一,和n是0,1,2或者3,例如甲氧基,和r4和r10的至少一个是在2'位置用烷基或者芳基如苯基取代的苯基。
在又一方面,r4和r10二者是在3'和5'位置用c1-c10烷基例如叔丁基取代的苯基。
在又一方面,r4和r10至少一个是在3'和5'位置用芳基例如取代或者未取代的苯基取代的苯基。
在又一方面,r4和r10二者是在3'和5'位置用芳基例如取代或者未取代的苯基取代的苯基。
在另一方面,r4和r10的至少一个是在3'和5'位置用c1-c10烷基(例如叔丁基,仲丁基,正丁基,异丙基,正丙基,环丙基,环丁基,环戊基,环己基,环庚基,环辛基,苯基,三甲基苯基或者金刚烷基)或者芳基及其组合取代的芳基,其中当r4或者r10是苯基(其进一步用芳基取代)时,该键合在一起的两个基团可以直接或者通过连接基团结合在一起,其中该连接基团是烷基,乙烯基,苯基,炔基,甲硅烷基,甲锗烷基,胺,铵,膦,鏻,醚,硫醚,硼烷,硼酸根,铝烷或者铝酸根基团。
供选择地,当r4和r10的至少一个是在3'和5'位置取代的苯基时,该苯基还可以在4'位置取代,优选用选自(xr’n)-的取代基取代,其中x是第14,15,16或者17族杂原子,其原子量是13-79(优选n,o,s,p或者si)和r’是氢原子,卤素原子,c1-c10烷基(例如甲基,乙基,丙基,丁基,戊基,己基,辛基,壬基,癸基或者其异构体),或者c6-c10芳基之一,和n是0,1,2或者3;优选(xr’n)-是–nr’2,-sr’,-or’,-osir’3,-sir’3或者-pr’2,优选(xr’n)-是–nr’2,–sr’,–or’,–osir’3或者–pr’2,优选(xr’n)-是–sr’,–or’或者–osir’3,优选(xr’n)-是–nr’2或者–pr’2,或者优选(xr’n)-是–or’m,优选其中r’是c1-c10烷基,特别是甲氧基,乙氧基,正丙氧基,异丙氧基,正丁氧基,异丁氧基,仲丁氧基或者叔丁氧基,最特别是甲氧基。
在再一方面,m是hf,ti和/或zr,特别是hf和/或zr,特别是zr。
用于基团r1,r3,r5,r6,r7,r9,r11,r12,r13和r14每个的合适的基团独立地选自氢或者烃基,包括甲基,乙基,乙烯基,以及丙基,丁基,戊基,己基,庚基,辛基,壬基,癸基,十一烷基,十二烷基,丙烯基,丁烯基的全部异构体(包括环状物例如环己基),和选自卤代碳基和卤代碳基的全部异构体,包括全氟丙基,全氟丁基,全氟乙基,全氟甲基,和选自取代的烃基和取代的烃基的全部异构体,包括三甲基甲硅烷基丙基,三甲基甲硅烷基甲基,三甲基甲硅烷基乙基,和选自苯基,和烃基取代的苯基的全部异构体,包括甲基苯基,二甲基苯基,三甲基苯基,四甲基苯基,五甲基苯基,二乙基苯基,三乙基苯基,丙基苯基,二丙基苯基,三丙基苯基,二甲基乙基苯基,二甲基丙基苯基,二甲基丁基苯基,二丙基甲基苯基等;选自卤素取代的苯基的全部异构体(其中卤素独立地是氟,氯,溴和碘),包括卤代苯基,二卤苯基,三卤苯基,四卤苯基和五卤苯基;和选自卤素取代的烃基取代的苯基的全部异构体(其中卤素独立地是氟,氯,溴和碘),包括卤甲基苯基,二卤甲基苯基,(三氟甲基)苯基,双(三氟甲基)苯基;和选自苄基的全部异构体,和烃基取代的苄基的全部异构体,包括甲基苄基,二甲基苄基。
在本发明的其他实施方案中,每个x独立地选自具有1-20个碳原子的烃基,氢基(hydride),胺基(amide),烷氧基,硫基,磷基,卤基,二烯,胺,膦,醚及其组合(两个x可以形成稠环或者环体系的一部分)。
x的合适的例子包括氯基,溴基,氟基,碘基,氢基和c1-c20烃基,优选甲基,乙基,丙基,丁基,戊基,己基,苯基,苄基及其全部异构体,或者两个x一起选自c4-c10二烯,优选丁二烯,甲基丁二烯,戊二烯,甲基戊二烯,二甲基戊二烯,己二烯,甲基己二烯,二甲基己二烯,或者选自c1-c10烷叉基,优选甲叉基,乙叉基,丙叉基,或者c3-c10烷基二基,优选丙二基,丁二基,戊二基和己二基。特别地,x是氯基或者甲基。
在任何实施方案中,t是选自下面的桥连基团:r’2c,r’2si,r’2ge,r’2ccr’2,r’2ccr’2cr’2,r’c=cr’,r’c=cr’cr’2,r’2csir’2,r’2sisir’2,r’2csir’2cr’2,r’2sicr’2sir’2,r’c=cr’sir’2,r’2cger’2,r’2geger’2,r’2cger’2cr’2,r’2gecr’2ger’2,r’2siger’2,r’c=cr’ger’2,r’b,r’2c–br’,r’2c–br’–cr’2,r’n,r’2c–nr’,r’2c–nr’–cr’2,r’p,r’2c–pr’和r’2c–pr’–cr’2,其中r’独立地是氢,烃基,取代的烃基,卤代碳基,取代的卤代碳基,甲硅烷基碳基或者甲锗烷基碳基,和相同原子或者相邻原子上的两个或者更多个r’可以结合在一起来形成取代或者未取代的,饱和的,部分不饱和的或者芳族环状或者多环取代基。
桥连基团t合适的例子包括二烃基亚甲硅烷基,包括二甲基亚甲硅烷基,二乙基亚甲硅烷基,二丙基亚甲硅烷基,二丁基亚甲硅烷基,二戊基亚甲硅烷基,二己基亚甲硅烷基,甲基苯基亚甲硅烷基,二苯基亚甲硅烷基,二环己基亚甲硅烷基,甲基环己基亚甲硅烷基,二苄基亚甲硅烷基,四甲基二亚甲硅烷基,环三亚甲基亚甲硅烷基,环四亚甲基亚甲硅烷基,环五亚甲基亚甲硅烷基,二乙烯基亚甲硅烷基,和四甲基二亚硅氧烷;二烃基亚甲锗烷基,包括二甲基亚甲锗烷基,二乙基亚甲锗烷基,二丙基亚甲锗烷基,二丁基亚甲锗烷基,甲基苯基亚甲锗烷基,二苯基亚甲锗烷基,二环己基亚甲锗烷基,甲基环己基亚甲锗烷基,环三亚甲基亚甲锗烷基,环四亚甲基亚甲锗烷基,和环五亚甲基亚甲锗烷基;亚二价碳基(carbylene)和二价碳基二基(carbdiyl),包括亚甲基,二甲基亚甲基,二乙基亚甲基,二丁基亚甲基,二丙基亚甲基,二苯基亚甲基,二甲苯基亚甲基,二(丁基苯基)亚甲基,二(三甲基甲硅烷基苯基)亚甲基,二苄基亚甲基,环四亚甲基亚甲基,环五亚甲基亚甲基,亚乙基,甲基亚乙基,二甲基亚乙基,三甲基亚乙基,四甲基亚乙基,环异丙基,环亚丁基,环亚戊基,环亚己基,丙二基,甲基丙二基,二甲基丙二基,三甲基丙二基,四甲基丙二基,五甲基丙二基,六甲基丙二基,亚乙烯基,和乙烯-1,1-二基;硼烷二基,包括甲基硼烷二基,乙基硼烷二基,丙基硼烷二基,丁基硼烷二基,戊基硼烷二基,己基硼烷二基,环己基硼烷二基和苯基硼烷二基;及其组合,包括二甲基甲硅烷基亚甲基,二苯基甲硅烷基亚甲基,二甲基甲硅烷基亚乙基,甲基苯基甲硅烷基亚甲基。
具体地,t是ch2,ch2ch2,c(ch3)2,sime2,siph2,simeph,si(ch2)3,si(ch2)4,si(me3siph)2,或者si(ch2)5。
在另一实施方案中,t是式r2aj所示的,其中j是c,si或者ge,和每个ra独立地是氢,卤素,c1-c20烃基或者c1-c20取代的烃基,和两个ra可以形成环状结构,包括芳族,部分饱和的,或者饱和的环状或者稠环体系。
在本发明的优选的实施方案中,在本文所述任何式中,t是式:(r*2g)g所示的,其中每个g是c,si或者ge,g是1或者2,和每个r*独立地是氢,卤素,c1-c20烃基或者c1-c20取代的烃基,和两个或者更多个r*可以形成环状结构,包括芳族,部分饱和的,或者饱和的环状或者稠环体系。
在本发明的方面,该茂金属催化剂的外消旋/内消旋之比是50:1或者更大,或者40:1或者更大,或者30:1或者更大,或者20:1或者更大,或者15:1或者更大,或者10:1或者更大,或者7:1或者更大,或者5:1或者更大。
在本发明的一实施方案中,该茂金属催化剂包含大于55mol%的外消旋异构体,或者大于60mol%的外消旋异构体,或者大于65mol%的外消旋异构体,或者大于70mol%的外消旋异构体,或者大于75mol%的外消旋异构体,或者大于80mol%的外消旋异构体,或者大于85mol%的外消旋异构体,或者大于90mol%的外消旋异构体,或者大于92mol%的外消旋异构体,或者大于95mol%的外消旋异构体,或者大于98mol%的外消旋异构体,基于外消旋和内消旋异构体(如何形成了任何的话)的总量。在本发明的一种具体实施方案中,茂金属,特别是桥连双(茚基)茂金属化合物基本上由外消旋异构体组成。
外消旋和内消旋异构体的量是通过质子nmr测定的。1hnmr数据是在23℃在5mm探针中使用400mhzbruker分光计,用氘化二氯甲烷或者氘化苯收集的。数据是使用最大脉冲宽度45°,脉冲间8秒和平均16个瞬态的信号来记录的。将所述光谱相对于氘化二氯甲烷中的质子化二氯甲烷进行标准化,其预期显示在5.32ppm的峰。
在优选的实施方案中,在本文所述的任何方法中,使用一种催化剂化合物,例如所述催化剂化合物不是不同的。就本发明的目的而言,如果一种茂金属化合物与另一种相差至少一个原子,则一种茂金属催化剂化合物被认为是与另一种不同的。例如,“联茚基二氯化锆”不同于“(茚基)(2-甲基茚基)二氯化锆”,其不同于“(茚基)(2-甲基茚基)二氯化铪”。仅仅异构体不同的催化剂化合物就本发明的目的而言被认为是相同的,例如外消旋-二甲基甲硅烷基双(2-甲基4-苯基)二甲基铪被认为与内消旋-二甲基甲硅烷基双(2-甲基4-苯基)二甲基铪相同。
在一些实施方案中,两种或者更多种不同的催化剂化合物存在于本文所用的催化剂体系中。在一些实施方案中,两种或者更多种不同的催化剂化合物存在于其中发生本文所述的方法(一种或多种)的反应区中。当两种基于过渡金属化合物的催化剂作为混合的催化剂体系用于一个反应器中时,这两种过渡金属化合物应当进行选择,以使得二者是相容性的。本领域技术人员已知的简单的筛选方法例如通过1h或者13cnmr筛选,可以用于确定那种过渡金属化合物是相容的。优选的是将相同的活化剂用于过渡金属化合物,然而,两种不同的活化剂例如非配位阴离子活化剂和铝氧烷可以组合使用。如果一种或多种过渡金属化合物包含x1或者x2配体(其不是氢基,烃基或者取代的烃基),则在加入非配位阴离子活化剂之前该铝氧烷通常与过渡金属化合物接触。
过渡金属化合物(预催化剂)可以以任何比率使用。(a)过渡金属化合物与(b)过渡金属化合物优选的摩尔比落入下面的范围:(a:b)1:1000-1000:1,供选择地1:100-500:1,供选择地1:10-200:1,供选择地1:1-100:1,和供选择地1:1-75:1,和供选择地5:1-50:1。所选择的具体比率将取决于所选择的确切的预催化剂,活化方法和期望的最终产物。在一种具体实施方案中,当使用两种预催化剂时(其中二者都是用相同的活化剂活化的),则有用的摩尔百分比(基于该预催化剂的分子量)是10-99.9%a比0.1-90%b,供选择地25-99%a比0.5-50%b,供选择地50-99%a比1-25%b,和供选择地75-99%a比1-10%b。
制备催化剂化合物的方法。
通常,这种类型的茂金属是如图13所示来合成的,其中(i)经由烷基阴离子的金属盐(例如nbuli)脱质子来形成茚化物(indenide);(ii)将茚化物与适当的桥连前体(例如me2sicl2)反应。(iii)上述产物与agotf反应。(iv)上述三氟甲基磺酸盐化合物与另一等价茚化物反应。(v)经由烷基阴离子(例如nbuli)双脱质子来形成二价阴离子;(vi)该二价阴离子与金属卤化物(例如zrcl4)反应。最终产物是通过粗固体的重结晶来获得的,其中ar1和ar2是如对于r4和r10所定义的,和r1和r2是如对于r2和r8所定义的。
在本文中有用的催化剂化合物包括:
me2si(4-oph2-2-nc6-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;ph2si(4-oph2-2-nc8-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-me-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-npr-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-npr-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-et-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-et-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-正丙基-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-nc3-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)(2-nc3-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nbu-ind)(2-npr-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc5-ind)(2-npr-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc7-ind)(2-正丙基-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc8-ind)(2-正丙基-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc9-ind)(2-nc3-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc10-ind)(2-正丙基-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-nc6-ind)2zrcl2;me2si(4-oph2-2-nc5-ind)2zrcl2;me2si(4-oph2-2-nbu-ind)2zrcl2;me2si(4-oph2-2-nc7-ind)2zrcl2;me2si2(4-oph2-2-nc8-ind)2zrcl2;me2si(4-oph2-2-nc9-ind)2zrcl2;me2si(4-oph2-2-nc10-ind)2zrcl2;me2si(2-nbu-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si(2-nc5-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si2(2-正己基-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si(2-nc7-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si(2-nc8-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si(2-nc9-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si(2-nc10-4-(3',5'-tbu2-4'-ome-ph)-ind)2zrcl2;me2si(2-nbu-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-nc5-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-正己基-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-nc7-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-nc8-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-nc9-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-nc10-4-(3',5'-tbu2ph)-ind)2zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-nbu-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-nc5-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-正己基-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-nc7-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-nc8-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-nc9-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2-4'-ome-ph)-ind)(2-nc10-4-(3',5'-tbu2-4'-ome-ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-nbu-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-nc5-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-正己基-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-nc7-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-nc8-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-nc9-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(2-me-4-(3',5'-tbu2ph)-ind)(2-nc10-4-(3',5'-tbu2ph)-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nbu-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nc5-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nc6-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nc7-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nc8-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nc9-ind)zrcl2;me2si(4-oph2-2-me-ind)(4-oph2-2-nc10-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nbu-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nc5-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nc6-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nc7-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nc8-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nc9-ind)zrcl2;me2si(4-oph2-2-et-ind)(4-oph2-2-nc10-ind)zrcl2;me2si(4-oph2-2-正丙基-ind)(4-oph2-2-nbu-ind)zrcl2;me2si(4-oph2-2-正丙基-ind)(4-oph2-2-nc5-ind)zrcl2;me2si(4-oph2-2-正丙基-ind)(4-oph2-2-nc6-ind)zrcl2;me2si(4-oph2-2-正丙基-ind)(4-oph2-2-nc7-ind)zrcl2;me2si(4-oph2-2-正丙基-ind)(4-oph2-2-nc8-ind)zrcl2;me2si(4-oph2-2-npr-ind))(4-oph2-2-nc9-ind)zrcl2;me2si(4-oph2-2-npr-ind)(4-oph2-2-nc10-ind)zrcl2;me2si(4-oph2-2-nc6-ind)2zrcl2;me2si(4-oph2-2-nc5-ind)2zrcl2;me2si(4-oph2-2-nbu-ind)2zrcl2;me2si(4-oph2-2-nc7-ind)2zrcl2;me2si(4-oph2-2-nc8-ind)2zrcl2;me2si(4-oph2-2-nc9-ind)2zrcl2;me2si(4-oph2-2-nc10-ind)2zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc6-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc7-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc8-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc9-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc10-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc6-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc7-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc8-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nc9-ind)(4-(3’,5’-tbu2-4’-omeph)-2-me-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc6-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc7-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc8-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc9-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc10-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc6-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc7-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc8-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nc9-ind)(4-(3’,5’-tbu2)-2-me-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2-4’-omeph)-2-et-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2-4’-omeph)-2-et-thi)zrcl2;me2si(4-oph2-2-nc6-ind)(4-(3’,5’-tbu2-4’-omeph)-2-et-thi)zrcl2;me2si(4-oph2-2-nc7-ind)(4-(3’,5’-tbu2-4’-omeph)-2-et-thi)zrcl2;me2si(4-oph2-2-nc8-ind)(4-(3’,5’-tbu2-4’-omeph)-2-et-thi)zrcl2;me2si(4-oph2-2-nc9-ind)(4-(3’,5’-tbu2-4’-omeph)-2-et-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正丙基-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正丙基-thi)zrcl2;me2si(4-oph2-2-nc6-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正丙基-thi)zrcl2;me2si(4-oph2-2-nc7-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正丙基-thi)zrcl2;me2si(4-oph2-2-nc8-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正丙基-thi)zrcl2;me2si(4-oph2-2-nc9-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正丙基-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nbu-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc5-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正己基-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc7-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc8-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc9-thi)zrcl2;me2si(4-oph2-2-me-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc10-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nbu-thi)zrcl2;me2si(4-oph2-2-nc5-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc5-thi)zrcl2;me2si(4-oph2-2-正己基-ind)(4-(3’,5’-tbu2-4’-omeph)-2-正己基-thi)zrcl2;me2si(4-oph2-2-nbu-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc7-thi)zrcl2;和me2si(4-oph2-2-正己基-ind)(4-(3’,5’-tbu2-4’-omeph)-2-nc8-thi)zrcl2,其中oph是邻苯基,nc6是正己基,t-bu2和tbu2是二叔丁基,nbu是正丁基,ome是甲氧基,ind是茚基,ph是苯基,nc3和npr是正丙基,oph2是邻联苯基,nc5是正戊基,nc7是正庚基,nc8是正辛基,nc9是正壬基,nc10是正癸基,me是甲基,et是乙基,thi是1,5,6,7-四氢对称引达省基,和ome-ph和omeph是甲氧基苯基。
活化剂
术语“助催化剂”和“活化剂”在本文中是可互换使用的,并且定义为是任何这样的化合物,其可以通过将中性催化剂化合物转化成催化活性的催化剂化合物阳离子,来活化上述催化剂化合物的任何一种。非限定性活化剂例如包括铝氧烷,烷基铝,离子化活化剂,其可以是中性或者离子的,和常规类型助催化剂。优选的活化剂通常包括铝氧烷化合物,改性的铝氧烷化合物,和离子化阴离子前体化合物,其夺取了反应性的,σ-键合的金属配体,使得金属络合物是阳离子的,并且提供电荷平衡的非配位或者弱配位的阴离子。
在一种实施方案中,铝氧烷活化剂作为活化剂用于催化剂组合物中。铝氧烷通常是低聚物化合物,其含有-al(r1)-o-子单元,其中r1是烷基。铝氧烷的例子包括甲基铝氧烷(mao),改性的甲基铝氧烷(mmao),乙基铝氧烷和异丁基铝氧烷。烷基铝氧烷和改性的烷基铝氧烷适于作为催化剂活化剂,特别是当可夺取配体是烷基,卤基,烷氧基或者胺基时更是如此。不同的铝氧烷和改性铝氧烷的混合物也可以使用。可以优选使用目视澄清的甲基铝氧烷。雾浊的或者凝胶化的铝氧烷可以过滤来生产澄清溶液或者澄清铝氧烷可以从雾浊溶液中滗析。有用的铝氧烷是改性的甲基铝氧烷(mmao)助催化剂类型3a(在商标名modifiedmethylalumoxane3a类型下可商购获自akzochemicals,inc.,其涵盖于专利号us5041584下)。
当该活化剂是铝氧烷(改性或者未改性的)时,一些实施方案选择最大量的活化剂,其为相对于催化剂化合物(预金属催化活性位置)5000倍摩尔过量的al/m。最小活化剂与催化剂化合物摩尔比是1:1。供选择地优选的范围包括1:1-1000:1,供选择地1:1-500:1,供选择地1:1-200:1,供选择地1:1-100:1,或者供选择地1:1-50:1。
在一种供选择地实施方案中,很少或者没有铝氧烷用于本文所述的聚合方法中。优选,铝氧烷存在量是0mol%,供选择地铝氧烷以铝与催化剂化合物过渡金属的摩尔比小于500:1,优选小于300:1,优选小于100:1,优选小于1:1存在。
术语“非配位阴离子”(nca)表示这样的阴离子,其没有配位到阳离子,或者其仅仅弱配位到阳离子,由此保留了足部的不稳定性来通过中性路易斯碱来置换。“相容性”非配位阴离子是那些,其在初始形成的络合物分解时不降解到中性。此外,该阴离子将不会使阴离子取代基或者片段转移到阳离子来使得它由阴离子形成中性过渡金属化合物和中性副产物。根据本发明可用的非配位阴离子是那些,其是与过渡金属阳离子相容的,并且在以+1平衡它的离子电荷的含义上稳定了后者,并且仍然保持了足够的不稳定性来允许在聚合过程中置换。
处于本发明范围内的是使用离子化或者化学计量活化剂,中性或者离子的,例如三(正丁基)铵四(五氟苯基)硼酸盐,三全氟苯基硼类金属前体或者三全氟萘基硼类金属前体,多卤代杂硼烷阴离子(wo98/43983),硼酸(us5942459)或者其组合。还处于本发明范围内的是使用单独的中性或者离子的活化剂或者其与铝氧烷或者改性铝氧烷活化剂的组合。
中性化学计量活化剂的例子包括三取代的硼,碲,铝,镓和铟或者其混合物。三种取代基每个独立地选自烷基,烯基,卤素,取代的烷基,芳基,芳基卤基,烷氧基和卤基。优选,该三个基团独立地选自卤素,单环或者多环(包括卤代)芳基,烷基,和烯基化合物及其混合物,优选的是具有1-20个碳原子的烯基,具有1-20个碳原子的烷基,具有1-20个碳原子的烷氧基和具有3-20个碳原子的芳基(包括取代的芳基)。更优选,该三个基团是具有1-4个碳基团的烷基,苯基,萘基或者其混合物。甚至更优选,该三个基团是卤代的,优选氟代的芳基。优选的中性化学计量活化剂是三全氟苯基硼或者三全氟萘基硼。
离子化学计量活化剂化合物可以包含活性质子,或者一些其他阳离子,其连接但不配位到,或者仅仅松散配位到所述离子化化合物的其余离子上。这样的化合物等描述在欧洲公开ep0570982a;ep0520732a;ep0495375a;ep0500944b1;ep0277003a;ep0277004a;us5153157;us5198401;us5066741;us5206197;us5241025;us5384299;us5502124;和ussn08/285380(1994年8月3日提交);其全部通过引用完全并入本文。
可用作本发明方法中的活化剂的优选的化合物包含阳离子(其优选是能够贡献质子的布朗斯台德酸),和相容性非配位阴离子,该阴离子是相对大的(大体积),能够稳定活性催化剂物质(第4族阳离子),其当两种化合物合并时形成,并且所述的阴离子将足够地不稳定来通过烯属,二烯属和炔属不饱和基物或者其他中性路易斯碱,例如醚,胺等来置换。两类有用的相容性非配位阴离子已经公开在ep0277003a1和ep0277004a1中:1)阴离子配位络合物,其包含多个亲脂性基团,其共价配位到和遮蔽了中心带电金属或者非金属核;和2)阴离子,其包含多个硼原子例如碳硼烷,金属碳硼烷和硼烷。
在一种优选的实施方案中,该化学计量活化剂包括阳离子和阴离子组分,和优选是下式(ii)所示的:
(z)d+(ad-)(ii)
其中:z是(l-h)或者可还原的路易斯酸;l是中性路易斯碱;h是氢;(l-h)+是布朗斯台德酸;ad-是具有电荷d-的非配位阴离子;d是1-3。
当z是(l-h)以使得阳离子组分是(l-h)d+时,该阳离子组分可以包括布朗斯台德酸例如质子化路易斯碱,其能够质子化来自于含有大体积配体茂金属的过渡金属催化剂前体的部分例如烷基或者芳基,形成阳离子过渡金属物质。优选,该活化阳离子(l-h)d+是布朗斯台德酸,其能够将质子贡献给过渡金属催化前体,产生过渡金属阳离子,包括铵,氧鎓,鏻,甲硅烷鎓及其混合物,优选来自于下面的铵:甲基胺,苯胺,二甲基胺,二乙基胺,n-甲基苯胺,二苯基胺,三甲基胺,三乙基胺,n,n-二甲基苯胺,甲基二苯基胺,吡啶,对溴n,n-二甲基苯胺,对硝基-n,n-二甲基苯胺,来自于下面的鏻:三乙基膦,三苯基膦和二苯基膦,来自于醚例如二甲基醚二乙醚,四氢呋喃和二氧六环的氧鎓,来自于硫醚例如二乙基硫醚和四氢噻吩的锍,及其混合物。
当z是可还原的路易斯酸时,它优选是式:(ar3c+)所示,其中ar是芳基或者用杂原子,c1-c40烃基或者取代的c1-c40烃基取代的芳基,优选该可还原路易斯酸是式:(ph3c+)所示的,其中ph是苯基或者用杂原子,c1-c40烃基或者取代的c1-c40烃基取代的苯基。在一种优选的实施方案中,该可还原路易斯酸是三苯基碳鎓。
阴离子组分ad-包括具有式[mk+qn]d-的那些,其中k是1,2或者3;n是1,2,3,4,5或者6,优选3,4,5或6;n-k=d;m是选自元素周期表第13族的元素,优选硼或者铝,和q独立地是氢基,桥连或者未桥连的二烷基氨基,卤基,烷氧基,芳氧基,烃基,取代的烃基,卤代碳基,取代的卤代碳基,和卤素取代的烃基,所述的q具有至多20个碳原子,并且条件是在不多于一次出现中q是卤基,并且两个q基团可以形成环结构。优选,每个q是具有1-20个碳原子的氟代烃基,更优选每个q是氟代芳基,和最优选每个q是五氟芳基。合适的ad-组分的例子还包括二硼化合物,如美国专利no.5447895所公开的,其通过引用完全并入本文。
在一种优选的实施方案中,本发明涉及聚合烯烃的方法,其包括将烯烃(优选乙烯)与催化剂化合物和式(14)所示的含硼的nca活化剂接触:
zd+(ad-)(14)
其中:z是(l-h)或者可还原路易斯酸;l是中性路易斯碱(如上面进一步所述的);h是氢;(l-h)是布朗斯台德酸(如上面进一步所述的);ad-是具有电荷d-的含硼的非配位阴离子(如上面进一步所述的);d是1,2或者3。
在一种优选的实施方案中,在上述式14所示的任何nca中,该可还原路易斯酸是式:(ar3c+)所示的,其中ar是芳基或者用杂原子,c1-c40烃基或者取代的c1-c40烃基取代的芳基,优选该可还原路易斯酸是式:(ph3c+)所示的,其中ph是苯基或者用杂原子,c1-c40烃基或者取代的c1-c40烃基取代的苯基。
在一种优选的实施方案中,在上述式14所示的任何nca中,zd+是式:(l-h)d+所示的,其中l是中性路易斯碱;h是氢;(l-h)是布朗斯台德酸;和d是1,2或者3,优选(l-h)d+是布朗斯台德酸,选自铵,氧鎓,鏻,甲硅烷鎓及其混合物。
在一种优选的实施方案中,在上述式14所示的任何nca中,阴离子组分ad-是式[m*k*+q*n*]d*-所示的,其中k*是1,2或者3;n*是1,2,3,4,5或者6(优选1,2,3或者4);n*-k*=d*;m*是硼;和q*独立地选自氢基,桥连或者未桥连的二烷基胺基,卤基,烷氧基,芳氧基,烃基,取代的烃基,卤代碳基,取代的卤代碳基和卤素取代的烃基,所述的q*具有至多20个碳原子,条件是在不多于一次出现中q*是卤基。
本发明还涉及聚合烯烃的方法,其包括将烯烃(例如乙烯)与催化剂化合物和式(i)所示的nca活化剂接触:
rnm**(arnhal)4-n(i)
其中r是单阴离子配体;m**是第13族金属或者类金属;arnhal是卤代的含氮的芳族环,多环芳族环,或者芳族环集合体,其中两个或者更多个环(或者稠环体系)是直接彼此结合或者共同结合的;和n是0,1,2或者3。通常,式i的含阴离子的nca还包含合适的阳离子,其基本上不干扰与过渡金属化合物形成的离子催化剂络合物,优选该阳离子是上述zd+。
在一种优选的实施方案中,在上述式i所示的任何含阴离子的nca中,r选自取代或者未取代的c1-c30烃基脂肪族或者芳族基团,其中取代表示碳原子上的至少一个氢是用下面的基团替代的:烃基,卤基,卤代碳基,烃基或者卤代碳基取代的有机类金属,二烷基氨基,烷氧基,芳氧基,烷基硫基,芳基硫基,烷基磷基,芳基磷基或者其他阴离子取代基;氟基;大体积烷氧基,其中大体积表示c4-c20烃基;--sr1,--nr22和--pr32,其中每个r1,r2或者r3独立地是上面定义的取代或者未取代的烃基;或者c1-c30烃基取代的有机类金属。
在一种优选的实施方案中,在上述式i所示的任何含阴离子的nca中,该nca还包含式:(ar3c+)所示的含阳离子的可还原路易斯酸,其中ar是芳基或者用杂原子,c1-c40烃基或者取代的c1-c40烃基取代的芳基,优选可还原路易斯酸是式:(ph3c+)所示,其中ph是苯基或者用杂原子,c1-c40烃基或者取代的c1-c40烃基取代的苯基。
在一种优选的实施方案中,在上述式i所示的任何含阴离子的nca中,该nca还包含式(l-h)d+所示的阳离子,其中l是中性路易斯碱;h是氢;(l-h)是布朗斯台德酸;和d是1,2或者3,优选(l-h)d+是布朗斯台德酸,其选自铵,氧鎓,鏻,甲硅烷鎓及其混合物。
有用的活化剂另外的例子包括公开在us7297653和7799879中的那些。
在本文中有用的另一活化剂包含式(16)所示的阳离子氧化剂和非配位相容性阴离子的盐:
(oxe+)d(ad-)e(16)
其中oxe+是具有电荷e+的阳离子氧化剂;e是1,2或者3;d是1,2或者3;和ad-是具有电荷d-的非配位阴离子(如上面进一步所述)。阳离子氧化剂的例子包括:二茂铁,烃基取代的二茂铁,ag+,或者pb+2。ad-优选的实施方案包括四(五氟苯基)硼酸根。
在另一实施方案中,本文所述的催化剂化合物可以与大体积活化剂一起使用。如本文所用的,“大体积活化剂”指的是下式所示的阴离子活化剂:
其中:
每个r1独立地是卤基,优选氟基;
每个r2独立地是卤基,c6-c20取代的芳族烃基或者式–o-si-ra的硅氧烷基团,其中ra是c1-c20烃基或者烃基甲硅烷基(优选r2是氟基或者全氟代苯基);
每个r3是卤基,c6-c20取代的芳族烃基或者式–o-si-ra的硅氧烷基团,其中ra是c1-c20烃基或者烃基甲硅烷基(优选r3是氟基或者c6全氟代芳族烃基);其中r2和r3可以形成一个或多个饱和或者不饱和的,取代或者未取代的环(优选r2和r3形成全氟代苯基环);
l是中性路易斯碱;(l-h)+是布朗斯台德酸;d是1,2或者3;
其中该阴离子的分子量大于1020g/mol;和
其中b原子上至少三个取代基每个的分子体积大于
“分子体积”在本文中用作活化剂分子在溶液中的空间立体体积的近似来。不同分子体积的取代基的比较使得与具有较大分子体积的取代基相比,具有较小分子体积的取代基被认为是“较小大体积的”。相反,与具有较小分子体积的取代基相比,具有较大分子体积的取代基可以被认为是“较大大体积的”。
分子体积可以如“asimple“backoftheenvelope”methodforestimatingthedensitiesandmolecularvolumesofliquidsandsolids,”journalofchemicaleducation,第71卷,第11期,1994年11月,第962-964页所报告来计算。分子体积(mv,单位是
适于本文的活化剂的示例性大体积取代基和它们各自的缩放体积和分子体积显示在us9266977第20栏第35行以后的表格中。
对于特别有用的大体积活化剂的列表,请参见us8658556,其通过引用并入本文。
在另一实施方案中,一种或多种的nca活化剂选自us6211105所述的活化剂。
优选的活化剂包括n,n-二甲基苯胺鎓四(全氟萘基)硼酸盐,n,n-二甲基苯胺鎓四(全氟联苯基)硼酸盐,n,n-二甲基苯胺鎓四(全氟苯基)硼酸盐,n,n-二甲基苯胺鎓四(3,5-双(三氟甲基)苯基)硼酸盐,三苯基碳鎓四(全氟萘基)硼酸盐,三苯基碳鎓四(全氟联苯基)硼酸盐,三苯基碳鎓四(3,5-双(三氟甲基)苯基)硼酸盐,三苯基碳鎓四(全氟苯基)硼酸盐,[ph3c+][b(c6f5)4-],[me3nh+][b(c6f5)4-];1-(4-(三(五氟苯基)硼酸根)-2,3,5,6-四氟苯基)吡咯烷鎓;和四(五氟苯基)硼酸盐,4-(三(五氟苯基)硼酸根)-2,3,5,6-四氟吡啶。
在一种优选的实施方案中,该活化剂包含三芳基碳鎓(例如三苯基碳鎓四苯基硼酸盐,三苯基碳鎓四(五氟苯基)硼酸盐,三苯基碳鎓四-(2,3,4,6-四氟苯基)硼酸盐,三苯基碳鎓四(全氟萘基)硼酸盐,三苯基碳鎓四(全氟联苯基)硼酸盐,三苯基碳鎓四(3,5-双(三氟甲基)苯基)硼酸盐)。
在另一实施方案中,该活化剂包含下面中的一种或多种:三烷基铵四(五氟苯基)硼酸盐,n,n-二烷基苯胺鎓四(五氟苯基)硼酸盐,n,n-二甲基-(2,4,6-三甲基苯胺鎓)四(五氟苯基)硼酸盐,三烷基铵四-(2,3,4,6-四氟苯基)硼酸盐,n,n-二烷基苯胺鎓四-(2,3,4,6-四氟苯基)硼酸盐,三烷基铵四(全氟萘基)硼酸盐,n,n-二烷基苯胺鎓四(全氟萘基)硼酸盐,三烷基铵四(全氟联苯基)硼酸盐,n,n-二烷基苯胺鎓四(全氟联苯基)硼酸盐,三烷基铵四(3,5-双(三氟甲基)苯基)硼酸盐,n,n-二烷基苯胺鎓四(3,5-双(三氟甲基)苯基)硼酸盐,n,n-二烷基-(2,4,6-三甲基苯胺鎓)四(3,5-双(三氟甲基)苯基)硼酸盐,二-(异丙基)铵四(五氟苯基)硼酸盐,(其中烷基是甲基,乙基,丙基,正丁基,仲丁基或者叔丁基)。
在一种优选的实施方案中,在与催化剂化合物组合之前或之后,优选在与催化剂化合物混合之前,任何本文所述的活化剂可以混合在一起。
在一些实施方案中,两种nca活化剂可以用于聚合中,并且第一nca活化剂与第二nca活化剂的摩尔比可以是任何比率。在一些实施方案中,第一nca活化剂与第二nca活化剂的摩尔比是0.01:1-10000:1,优选0.1:1-1000:1,优选1:1-100:1。
此外,通常的活化剂与催化剂之比,例如全部nca活化剂与催化剂之比是1:1摩尔比。供选择的优选的范围包括0.1:1-100:1,供选择地0.5:1-200:1,供选择地1:1-500:1,供选择地1:1-1000:1。特别有用的范围是0.5:1-10:1,优选1:1-5:1。
还处于本发明范围的是催化剂化合物可以与铝氧烷和nca的组合物合并(参见例如us5153157,us5453410,ep0573120b1,wo94/07928和wo95/14044,其讨论了铝氧烷与离子化活化剂组合使用)。
任选的清除剂或者助活化剂
除了这些活化剂化合物之外,还可以使用清除剂或者助活化剂。烷基铝或者有机铝化合物(其可以用作清除剂或者助活化剂)包括例如三甲基铝,三乙基铝,三异丁基铝,三正己基铝,三正辛基铝和二乙基锌。
链转移剂
本发明进一步涉及在链转移剂存在下使用上述催化剂聚合烯烃的方法。
“链转移剂”是任何这样的试剂,其能够在聚合过程中在配位聚合催化剂和该链转移剂的金属中心之间进行烃基和/或聚合物基团交换。链转移剂可以是任何令人期望的化学化合物例如公开在wo2007/130306中的那些。优选,该链转移剂选自第2,12或者13族烷基或者芳基化合物;优选烷基或者芳基锌、镁或者铝;优选其中该烷基是c1-c30烷基,供选择地c2-c20烷基,供选择地c3-12烷基,通常独立地选自甲基,乙基,丙基,丁基,异丁基,叔丁基,戊基,己基,环己基,苯基,辛基,壬基,癸基,十一烷基和十二烷基;和其中二乙基锌是特别优选的。
在一种特别有用的实施方案中,本发明涉及催化剂体系,其包含活化剂,本文所述的催化剂化合物和链转移剂,其中该链转移剂选自第2,12或者13族烷基或者芳基化合物。
在一种特别有用的实施方案中,该链转移剂选自二烷基锌化合物,其中该烷基独立地选自甲基,乙基,丙基,丁基,异丁基,叔丁基,戊基,己基,环己基和苯基。
在一种特别有用的实施方案中,该链转移剂选自三烷基铝化合物,其中该烷基独立地选自甲基,乙基,丙基,丁基,异丁基,叔丁基,戊基,己基,环己基和苯基。
有用的链转移剂通常的存在量是10或者20或者50或者100当量到600或者700或者800或者1000当量,相对于催化剂组分。供选择地,该链转移剂(“cta”)是以催化剂络合物与cta摩尔比是大约1:3000-10:1;供选择地1:2000-10:1;供选择地1:1000-10:1;供选择地1:500-1:1;供选择地1:300-1:1;供选择地1:200-1:1;供选择地1:100-1:1;供选择地1:50-1:1;供选择地1:10-1:1存在的。
有用的链转移剂包括二乙基锌,三正辛基铝,三甲基铝,三乙基铝,三异丁基铝,三正己基铝,二乙基氯化铝,二丁基锌,二正丙基锌,二正己基锌,二正戊基锌,二正癸基锌,二正十二烷基锌,二正十四烷基锌,二正十六烷基锌,二正十八烷基锌,二苯基锌,二异丁基氢化铝,二乙基氢化铝,二正辛基氢化铝,二丁基镁,二乙基镁,二己基镁和三乙基硼。
任选的载体材料
在本文的实施方案中,所述催化剂体系可以包含惰性载体材料。优选该负载的材料是多孔载体材料例如滑石,和无机氧化物。其他载体材料包括沸石,粘土,有机粘土或者任何其他有机或者无机载体材料等,或者其混合物。
优选,该载体材料是细分形式的无机氧化物。用于本文的茂金属催化剂体系中的合适的无机氧化物材料包括第2,4,13和14族金属氧化物,例如二氧化硅,氧化铝及其混合物。可以单独或者与二氧化硅或氧化铝组合使用的其他无机氧化物是氧化镁,二氧化钛,氧化锆等。然而,可以使用其他合适的载体材料,例如细分的功能化聚烯烃,例如细分的聚乙烯。特别有用的载体包括氧化镁,二氧化钛,氧化锆,蒙脱石,页硅酸盐,沸石,滑石,粘土等。此外,可以使用这些载体材料的组合,例如二氧化硅-铬,二氧化硅-氧化铝,二氧化硅-二氧化钛等。优选的载体材料包括al2o3,zro2,sio2及其组合,更优选sio2,al2o3或者sio2/al2o3。
优选的是所述载体材料,最优选无机氧化物,表面积是大约10-大约700m2/g,孔体积是大约0.1-大约4.0cc/g和平均粒度是大约5-大约500μm。更优选,该载体材料的表面积是大约50-大约500m2/g,孔体积是大约0.5-大约3.5cc/g和平均粒度是大约10-大约200μm。最优选该载体材料的表面积是大约100-大约400m2/g,孔体积是大约0.8-大约3.0cc/g和平均粒度是大约5-大约100μm。可用于本发明的载体材料的平均孔尺寸是
所述载体材料应当是干燥的,即,没有吸收的水。载体材料的干燥可以通过在大约100℃-大约1000℃,优选至少大约600℃加热或煅烧来实现。当载体材料是二氧化硅时,将它加热到至少200℃,优选大约200℃-大约850℃和最优选大约600℃;并且进行大约1分钟-大约100小时,大约12小时-大约72小时,或者大约24小时-大约60小时的时间。该煅烧的载体材料必须具有至少一些反应性羟基(oh)来生产本发明的负载的催化剂体系。该煅烧的载体材料然后与包含至少一种茂金属化合物和活化剂的至少一种聚合催化剂接触。
所述具有反应性表面基团(通常是羟基)的载体材料在非极性溶剂中制浆,并且将所形成的淤浆与茂金属化合物和活化剂的溶液接触。在一些实施方案中,该载体材料的淤浆首先与活化剂接触大约0.5小时-大约24小时,大约2小时-大约16小时,或者大约4小时-大约8小时的时间。该茂金属化合物的溶液然后与分离的载体/活化剂接触。在一些实施方案中,该负载的催化剂体系是原位产生的。在供选择的实施方案中,该载体材料的淤浆首先与催化剂化合物接触大约0.5小时-大约24小时,大约2小时-大约16小时,或者大约4小时-大约8小时的时间。该负载的茂金属化合物的淤浆然后与活化剂溶液接触。
将该茂金属、活化剂和载体的混合物加热到大约0℃-大约70℃,优选大约23℃-大约60℃,优选在室温。接触时间通常是大约0.5小时-大约24小时,大约2小时-大约16小时,或者大约4小时-大约8小时。
合适的非极性溶剂是这样的材料,其中本文所用的全部反应物,即,活化剂和茂金属化合物,是至少部分地可溶的,并且其在反应温度是液体。优选的非极性溶剂是烷烃,例如异戊烷,己烷,正庚烷,辛烷,壬烷和癸烷,虽然也可以使用各种其他材料,包括环烷烃,例如环己烷,芳烃例如苯,甲苯和乙苯。
聚合方法
在本文的实施方案中,本发明涉及聚合方法,其中单体(例如丙烯)和任选的共聚单体与催化剂体系接触,该催化剂体系包含上述活化剂和至少一种茂金属化合物。所述催化剂化合物和活化剂可以以任何次序合并,并且通常在与单体接触前合并。
在本文中有用的单体包括取代的或者未取代的c2-c40α烯烃,优选c2-c20α烯烃,优选c2-c12α烯烃,优选乙烯,丙烯,丁烯,戊烯,己烯,庚烯,辛烯,壬烯,癸烯,十一碳烯,十二碳烯及其异构体。在本发明的一种优选的实施方案中,所述单体包含丙烯和任选的共聚单体,其包含下面中的一种或多种:乙烯或者c4-c40烯烃,优选c4-c20烯烃,或者优选c6-c12烯烃。该c4-c40烯烃单体可以是线性的、支化的或者环状的。该c4-c40环状的烯烃可以是张紧或者未张紧的,单环的或者多环的,和可以任选的包括杂原子和/或一个或多个官能团。在另一优选的实施方案中,所述单体包含乙烯和任选的共聚单体,其包含下面中的一种或多种:c3-c40烯烃,优选c4-c20烯烃,或者优选c6-c12烯烃。该c3-c40烯烃单体可以是线性的、支化的或者环状的。该c3-c40环状的烯烃可以是张紧或者未张紧的,单环的或者多环的,和可以任选的包括杂原子和/或一个或多个官能团。
示例性的c2-c40烯烃单体和任选的共聚单体包括乙烯,丙烯,丁烯,戊烯,己烯,庚烯,辛烯,壬烯,癸烯,十一碳烯,十二碳烯,降冰片烯,降冰片二烯,二环戊二烯,环戊烯,环庚烯,环辛烯,环辛二烯,环十二碳烯,7-氧杂降冰片烯,7-氧杂降冰片二烯,其取代的衍生物及其异构体,优选己烯,庚烯,辛烯,壬烯,癸烯,十二碳烯,环辛烯,1,5-环辛二烯,1-羟基-4-环辛烯,1-乙酰氧基-4-环辛烯,5-甲基环戊烯,环戊烯,二环戊二烯,降冰片烯,降冰片二烯和它们各自的类似物和衍生物,优选降冰片烯,降冰片二烯和二环戊二烯。
本发明的聚合方法可以以本领域已知的任何方式来进行。可以使用本领域已知的任何悬浮,均相,本体,溶液(包括超临界),淤浆或者气相聚合方法。这样的方法可以以间歇,半间歇或者连续模式来进行。均相聚合方法和淤浆方法是优选的。(均相聚合方法定义为这样的方法,其中至少90wt%的产物在反应介质中是可溶的)。本体均相方法是特别优选的。(本体方法通常是这样的方法,其中到反应器的全部供料中单体浓度是70vol%或者更大)。供选择地,不存在溶剂或者稀释剂或者不加入溶剂或者稀释剂到反应介质中,(除了少量用作催化剂体系或者其他添加剂的载体,或者通常与单体一起的量;例如丙烯中的丙烷)。在另一实施方案中,所述方法是淤浆方法。如本文所用的,术语“淤浆聚合方法”表示这样的聚合方法,其中使用负载的催化剂,并且单体是在负载的催化剂颗粒上聚合的。至少95wt%的衍生自负载的催化剂的聚合物产物处于丸粒形式如固体颗粒(没有溶解在稀释剂中)。
用于聚合的合适的稀释剂/溶剂包括非配位惰性液体。例子包括直链和支链烃,例如异丁烷,丁烷,戊烷,异戊烷,己烷,异己烷,庚烷,辛烷,十二烷及其混合物;环状和脂环烃,例如环己烷,环庚烷,甲基环己烷,甲基环庚烷及其混合物,例如可以是商购获得的(isopartm);全卤化烃例如全氟c4-10烷烃,氯苯和芳族和烷基取代的芳族化合物,例如苯,甲苯,三甲基苯和二甲苯。合适的溶剂还包括液体烯烃,其可以充当单体或者共聚单体,包括乙烯,丙烯,1-丁烯,1-己烯,1-戊烯,3-甲基-1-戊烯,4-甲基-1-戊烯,1-辛烯,1-癸烯及其混合物。在一种优选的实施方案中,脂肪族烃溶剂用作溶剂,例如异丁烷,丁烷,戊烷,异戊烷,己烷,异己烷,庚烷,辛烷,十二烷及其混合物;环状和脂环烃,例如环己烷,环庚烷,甲基环己烷,甲基环庚烷及其混合物。在另一实施方案中,所述溶剂不是芳族化合物,优选芳族化合物在溶剂中的存在量小于1wt%,优选小于0.5wt%,优选小于0wt%,基于溶剂的重量。
在一种优选的实施方案中,用于聚合的单体和共聚单体的供料浓度是60vol%溶剂或更低,优选40vol%或更低,或者优选20vol%或更低,基于供料流的总体积。优选所述聚合是以本体方法进行的。
优选的聚合可以在适于获得期望的乙烯聚合物的任何温度和/或压力进行。通常的温度和/或压力包括大约0℃-大约300℃,优选大约20℃-大约200℃,优选大约35℃-大约150℃,优选大约40℃-大约120℃,优选大约70℃-大约120℃,优选大约80℃-大约120℃,优选大约90℃-大约120℃,优选大约95℃-大约110℃的温度;和大约0.35mpa-大约10mpa,优选大约0.45mpa-大约6mpa,或者优选大约0.5mpa-大约4mpa的压力。
在通常的聚合中,所述反应的运行时间是至多300分钟,优选大约5-250分钟,或者优选大约10-120分钟。
在本发明的另一实施方案中,该聚合温度优选是大约70℃-大约130℃,优选大约80℃-大约125℃,优选大约90℃-大约120℃,优选大约95℃-大约110℃和该聚合方法是均相方法,优选溶液方法。
在一些实施方案中,氢气在聚合反应器中以0.001-50psig(0.007-345kpa),优选0.01-25psig(0.07-172kpa),更优选0.1-10psig(0.7-70kpa)的分压存在。在一些实施方案中,氢气不加入该聚合反应器中,即,氢气可以来自于其他源存在,例如氢气产生催化剂,但是不加入反应器中。
在本发明的一种实施方案中,该催化剂活性是至少50g/mmol/h,优选500g/mmol/h或者更大,优选5000g/mmol/h或者更大,优选50000g/mmol/h或者更大,优选100000g/mmol/h或者更大,优选150000g/mmol/h或者更大,优选200000g/mmol/h或者更大,优选250000g/mmol/h或者更大,优选300000g/mmol/h或者更大,优选350000g/mmol/h或者更大。在一种供选择的实施方案中,烯烃单体的转化率是至少10%,基于聚合物收率和进入反应区的单体重量,优选20%或者更大,优选30%或者更大,优选50%或者更大,优选80%或者更大。
在一种优选的实施方案中,很少或者没有清除剂用于生产乙烯聚合物的方法中。优选,清除剂(例如三烷基铝)的存在量是0mol%,供选择地该清除剂是以清除剂金属与过渡金属摩尔比小于100:1,优选小于50:1,优选小于15:1,优选小于10:1而存在。
在一种优选的实施方案中,所述聚合:1)是在0-300℃(优选90-150℃,优选95-120℃)的温度进行的;2)是在大气压到10mpa(优选0.35-10mpa,优选0.45-6mpa,优选0.5-4mpa)的压力进行的;3)是在脂肪族烃溶剂(例如异丁烷,丁烷,戊烷,异戊烷,己烷,异己烷,庚烷,辛烷,十二烷及其混合物;环状和脂环烃,例如环己烷,环庚烷,甲基环己烷,甲基环庚烷及其混合物;优选其中芳族化合物优选在溶剂中的存在量小于1wt%,优选小于0.5wt%,优选是0wt%,基于溶剂的重量)中进行的;4)所述聚合优选在一个反应区中进行;5)该催化剂化合物的生产率是至少80000g/mmol/h(优选至少150000g/mmol/h,优选至少200000g/mmol/h,优选至少250000g/mmol/h,优选至少300000g/mmol/h);6)任选地不存在清除剂(例如三烷基铝化合物)(例如存在量是0mol%,供选择地清除剂是以清除剂金属与过渡金属摩尔比小于100:1,优选小于50:1,优选小于15:1,优选小于10:1而存在);和7)任选地,氢气在聚合反应器中以分压0.001-50psig(0.007-345kpa)(优选0.01-25psig(0.07-172kpa),更优选0.1-10psig(0.7-70kpa))存在。在一种优选的实施方案中,聚合所用的催化剂体系包含不大于一种催化剂化合物。“反应区”也称作“聚合区”,其是在其中进行聚合的容器,例如间歇式反应器。当多个反应器以串联或者并联构造使用时,每个反应器被认为是分别的聚合区。对于在间歇式反应器和连续反应器二者中的多级聚合,每个聚合级被认为是分别的聚合区。在一种优选的实施方案中,所述聚合在一个反应区中发生。室温是23℃,除非另有指示。
其他添加剂也可以根据需要用于聚合中,例如一种或多种清除剂,促进剂,改性剂,链转移剂(例如二乙基锌),还原剂,氧化剂,氢气,烷基铝或者硅烷。
在特别有用的实施方案中,本发明涉及聚合烯烃的方法,其包括:1)在60℃或者更大(供选择地80℃或者更大,供选择地90℃或者更大,供选择地95℃或者更大)的温度,将一种或多种烯烃与本文所述的催化剂体系接触(优选该催化剂化合物是以外消旋:内消旋之比为至少7:1存在);和2)获得聚合物,其g'vis是0.97或者更小(供选择地0.95或者更小,供选择地0.90或者更小)和mw是200000g/mol或者更大,其是通过gpc-dri测定的(供选择地300000g/mol或者更大,供选择地400000g/mol或者更大)。
优选,所述方法是在大约90℃-大约200℃的温度,大约0.35mpa-大约10mpa的压力,和以至多300分钟的时间在溶液、淤浆或者气体相中进行。
优选当通过本文的方法生产的聚合物的乙烯含量是10%时,则该聚合物具有这样的mw,该mw≥0.9倍的在相同聚合条件下(除了乙烯和丙烯单体浓度)使用相同催化剂体系所生产的,具有40wt%乙烯的丙烯乙烯共聚物的mw,并且两个mw都是200000g/mol(gpc-dri)或者更大。
在一种特别优选的实施方案,本发明涉及聚合烯烃的方法,其包括:1)在95℃或者更高的温度,将乙烯和丙烯与本文所述的催化剂体系接触;2)获得聚合物,其具有:a)0-20重量%乙烯,基于共聚物的重量;b)mw是50000g/mol或者更大(优选200000g/mol或者更大),其是通过gpc-dri测定的;c)熔点是x℃或者更大,其中x=(在相同条件下所制造的聚合物的tm,除了聚合温度是70℃)减去10℃,和任选地该聚合物具有这样的mw,该mw≥0.9倍的在相同聚合条件下(除了乙烯和丙烯单体浓度)使用相同催化剂体系所生产的,具有40wt%乙烯的丙烯乙烯共聚物的mw,该具有40wt%乙烯的丙烯乙烯共聚物的mw是200000g/mol(gpc-dri)或者更大。
气相聚合
通常,在用于生产聚合物的流化气相方法中,将含有一种或多种单体的气态流在催化剂存在下在反应性条件下连续循环穿过流化床。将该气态流从流化床抽出和再循环回到反应器中。同时,将聚合物产物从该反应器抽出和将新鲜单体加入来置换聚合的单体。示例性的气相聚合方法可以是如us4543399;us4588790;us5028670;us5317036;us5352749;us5405922;us5436304;us5453471;us5462999;us5616661;和us5668228中所讨论和所述的。
气相方法中的反应器压力可以在大约69kpa-大约3450kpa,大约690kpa-大约3450kpa,大约1380kpa-大约2759kpa,或者大约1724kpa-大约2414kpa变化。
该气相方法中的反应器温度可以在大约30℃-大约120℃,优选大约60℃-大约115℃,更优选大约65℃-110℃和最优选大约70℃-大约95℃变化。在另一实施方案中,当期望高密度聚乙烯时,该反应器温度通常是大约70℃-大约105℃。
气相体系中的催化剂或者催化剂体系的生产率受到主要单体分压的影响。主要单体乙烯或者丙烯,优选乙烯优选的摩尔百分比是大约25mol%-大约90mol%和共聚单体分压是大约138kpa-大约5000kpa,优选大约517kpa-大约2069kpa,其是气相聚合方法中通常的条件。此外在一些体系中,共聚单体的存在可以增加生产率。
在一种优选的实施方案中,所述反应器可以能够生产大于227kg聚合物/小时(kg/h)-大约90900kg/h或者更高,优选大于455kg/h,更优选大于4540kg/h,甚至更优选大于11300kg/h,又更优选大于15900kg/h,又甚至更优选大于22700kg/h和优选大于29000kg/h-大于45500kg/h,和最优选超过45500kg/h。
搅拌床中的聚合可以在一个或者两个水平搅拌容器中根据聚合模式来进行。所述反应器可以再分成单个气体组成可控和/或聚合温度可控的聚合隔间。基本上在反应器的一端处连续注入催化剂,和在另一端处除去粉末,停留时间分布接近于栓塞流反应器。优选氟碳化合物(如果存在的话)引入第一搅拌容器中。
本文所讨论和所述的方法所预期的其他气相方法可以包括描述在us5627242;us5665818;us5677375;ep-a-0794200;ep-a-0802202;和ep-b-634421中的那些。
在另一优选的实施方案中,所述催化剂体系处于液体,悬浮液,分散体和/或淤浆形式,并且可以引入气相反应器中进入树脂颗粒贫含区。将液体,悬浮液,分散体和/或淤浆催化剂体系引入流化床聚合中进入颗粒贫含区可以如us5693727中所讨论和所述。
在一些实施方案中,该气相聚合可以在不存在氟碳化合物时进行。在一些实施方案中,该气相聚合可以在氟碳化合物存在下进行。通常来说,氟碳化合物可以用作聚合介质和/或冷凝剂。
淤浆相聚合
淤浆聚合方法通常在大约103kpa-大约5068kpa或者甚至更大的压力范围和大约0℃-大约120℃的温度进行。在淤浆聚合中,在液体聚合稀释剂介质中形成了固态颗粒聚合物的悬浮液,向其中加入单体和共聚单体以及催化剂。该包括稀释剂的悬浮液是从反应器中间歇或者连续除去的,其中将挥发性组分与聚合物分离和再循环(任选地在蒸馏后)到反应器。聚合介质中所采用液体稀释剂通常是具有大约3-大约7个碳原子的烷烃介质,优选支化的烷烃。所用介质在聚合条件下可以是液体和相对惰性的。当使用丙烷介质时,所述方法可以高于反应稀释剂临界温度和压力来运行。优选,使用己烷或者异丁烷介质。
在一种实施方案中,优选的聚合技术(称作颗粒形成聚合或者淤浆方法)可以包括将温度保持低于聚合物进入溶液时的温度。这样的技术是本领域公知的,并且可以如us3248179中所讨论和所述。颗粒形成方法中优选的温度可以是大约20℃-大约110℃。用于淤浆方法的两种优选的聚合方法可以包括使用环路反应器的那些和利用串联、并联或者其组合的多个搅拌反应器的那些。淤浆方法的非限定性例子包括连续环路或者搅拌釜方法。此外,淤浆方法的其他例子可以如us4613484中所讨论和所述。
在另一实施方案中,该淤浆方法可以在环路反应器中连续进行。所述催化剂,作为在矿物油和/或链烷烃中的淤浆或者作为干燥的自由流动粉末,可以定期注入反应器环路中,其可以用在含有单体和共聚单体的稀释剂中增长的聚合物颗粒的循环淤浆来填充。氢气任选地可以作为分子量控制剂加入。反应器可以在大约3620kpa-大约4309kpa的压力和大约60℃-大约115℃的温度操作,这取决于期望的聚合物熔融特性。反应热可以通过环路壁除去,因为大部分反应器处于双套管的形式。使得所述淤浆定期或者连续离开该反应器依次到加热的低压闪蒸容器,旋转干燥器和氮气吹扫柱,来除去稀释剂和至少一部分任何未反应的单体和/或共聚单体。所形成的烃自由流动粉末可以复合来用于各种应用。
淤浆方法所用反应器可以产生大于907kg/h,更优选大于2268kg/h和最优选大于4540kg/h的聚合物。在另一实施方案中,该淤浆反应器可以产生大于6804kg/h,优选大于11340kg/h-大约45500kg/h。该淤浆方法所用反应器可以处于大约2758kpa-大约5516kpa,优选大约3103kpa-大约4827kpa,更优选大约3448kpa-大约4482kpa,最优选大约3620kpa-大约4309kpa的压力。
该淤浆方法的反应器液体介质中占优单体的浓度可以是大约1wt%-大约30wt%,优选大约2wt%-大约15wt%,更优选大约2.5wt%-大约10wt%,最优选大约3wt%-大约20wt%。
在一种或多种实施方案中,所述淤浆和/或气体相聚合可以在不存在或者基本上没有任何清除剂(例如三乙基铝,三甲基铝,三异丁基铝和三正己基铝和二乙基氯化铝,二丁基锌等)时操作。不存在或者基本上没有任何清除剂时操作淤浆和/或气体相反应器可以如wo96/08520和us5712352中所讨论和所述。在另一实施方案中,该聚合方法可以在具有清除剂的情况下来操作。通常的清除剂包括三甲基铝,三乙基铝,三异丁基铝,三正辛基铝和过量的铝氧烷和/或改性铝氧烷。
在一些实施方案中,该淤浆相聚合可以在不存在氟碳化合物时进行。在一些实施方案中,该淤浆相聚合可以在氟碳化合物存在下进行。通常来说,氟碳化合物可以用作聚合介质。
溶液相聚合
如本文所用的,措词“溶液相聚合”指的是这样的聚合体系,其中所生产的聚合物可溶于聚合介质。通常这包括在连续反应器中的聚合,在其中将所形成的聚合物和所供给的起始单体和催化剂材料搅拌来降低或者避免浓度梯度和在其中单体充当了稀释剂或者溶剂或者在其中烃用作稀释剂或者溶剂。合适的方法通常在大约0℃-大约250℃,优选大约50℃-大约200℃,优选大约80℃-大约150℃,更优选大约90℃-大约140℃,更优选大约95℃-大约120℃的温度和大约0.1mpa或者更大,优选2mpa或者更大的压力运行。对压力上限没有严格限制,但是通常可以是大约200mpa或者更小,优选120mpa或者更小。反应器中的温度控制通常可以通过平衡聚合热和使用反应器冷却(通过反应器套管或者冷却盘管来冷却反应器内容物,自动致冷,预冷却供料,液体介质(稀释剂、单体或者溶剂)的气化或者全部三种的组合)来获得。还可以使用具有预冷却供料的绝热反应器。对于特定类型的聚合的最大催化剂生产率,可以优化溶剂的纯度、类型和量。所述溶剂还可以作为催化剂载体引入。该溶剂可以作为气相或者液相引入,这取决于压力和温度。有利地,该溶剂可以保持在液体相和作为液体引入。溶剂可以在供料中引入聚合反应器。
在一种优选的实施方案中,所述聚合方法可以描述为连续的,非间歇方法,其在它的稳态操作时是通过单位时间所制造的聚合物的除去量基本上等于单位时间从反应容器抽出的聚合物的量来示例的。我们用“基本上等于”意指单位时间所制造的聚合物和单位时间所抽出的聚合物的这些量相互的比率是0.9:1;或者0.95:1;或者0.97:1;或者1:1。在这样的反应器中,将存在基本上均匀的单体分布。
优选在连续方法中,催化剂和聚合物在反应器中的平均停留时间通常可以是大约5分钟-大约8小时,和优选大约10分钟-大约6小时,更优选10分钟-1小时。在一些实施方案中,共聚单体(例如乙烯)可以以一定量加入反应容器中,来保持压力差超过主要单体(例如丙烯)和所存在的任何任选的二烯单体的合计蒸气压。
在另一实施方案中,所述聚合方法可以在乙烯压力为大约68kpa-大约6800kpa,最优选大约272-大约5440kpa进行。该聚合通常在大约25℃-大约250℃,优选大约75℃-大约200℃和最优选大约95℃-大约200℃的温度进行。
将少量烃加入通常的溶液相方法会引起聚合物溶液粘度下降和或聚合物溶质的量增加。在传统溶液方法中加入较大量的溶剂会引起聚合物分离成分别的相(其可以是固体或者液体,这取决于反应条件,例如温度或者压力)。
本文所讨论和所述的方法可以在连续搅拌釜反应器,间歇式反应器或者栓塞流反应器中进行。即使要进行顺序聚合时也可以使用一个反应器,优选只要在两个反应的时间或者空间中存在分隔就行。同样还可以使用两个或者更多个串联或并联操作的反应器。这些反应器可以具有或者不具有内冷却,和所述单体供料可以或者可以不被致冷。参见us5001205的关于一般的工艺条件的公开内容。还参见wo96/33227和wo97/22639。
超临界或者超溶液(supersolution)聚合
在本发明的方面,本文公开的方法和或催化剂组合物可以用于超临界或者超溶液相。超临界聚合表示这样的聚合方法,其中聚合体系是处于致密流体(即,它的密度是300kg/m3或者更高),超临界态。术语“致密流体”和“超临界态”是在us7812104中定义的。超溶液聚合是这样的聚合方法,其中聚合在65℃-150℃,优选大约75℃-大约140℃,优选大约90℃-大约140℃,更优选大约100℃-大约140℃的温度和1.72mpa-35mpa,优选5-30mpa的压力进行。关于超临界和超溶液聚合进一步的信息,请参见us7812104;us8008412;us7812104;us9249239;us7729536;us8058371;和us2008/0153997。
聚烯烃产物
本发明还涉及本文所述方法生产的物质的组合物。
在一种优选的实施方案中,本文所述的方法生产了丙烯均聚物或者丙烯共聚物,例如丙烯-乙烯和/或丙烯-α烯烃(优选c3-c20)共聚物(例如丙烯-己烯共聚物或者丙烯-辛烯共聚物),优选具有:mw/mn大于1-4(优选大于1-3)。
同样,本发明的方法生产了烯烃聚合物,优选聚乙烯和聚丙烯均聚物和共聚物。在一种优选的实施方案中,本文生产的聚合物是乙烯或丙烯的均聚物,或者是乙烯的共聚物,其优选具有0-50mol%(供选择地0.5-25mol%,供选择地0.5-20mol%,供选择地1-15mol%,优选3-10mol%)的一种或多种c3-c20烯烃共聚单体(优选c3-c12α-烯烃,优选丙烯,丁烯,己烯,辛烯,癸烯,十二碳烯,优选丙烯,丁烯,己烯,辛烯),或者是丙烯的共聚物,其优选具有0-25mol%(供选择地0.5-20mol%,供选择地1-15mol%,优选3-10mol%)的一种或多种c2或者c4-c20烯烃共聚单体(优选乙烯或者c4-c12α-烯烃,优选乙烯,丁烯,己烯,辛烯,癸烯,十二碳烯,优选乙烯,丁烯,己烯,辛烯)。
在一种优选的实施方案中,所述单体是乙烯和所述共聚单体是己烯,优选1-15mol%己烯,供选择地1-10mol%。
在一种优选的实施方案中,所述单体是丙烯和所述共聚单体是乙烯,优选0.5-99.5wt%乙烯,供选择地1-65wt%乙烯,供选择地1-60wt%乙烯,供选择地2-50wt%乙烯,供选择地3-30wt%乙烯,供选择地4-20wt%乙烯,基于该共聚物的重量。
通常,本文所生产的聚合物的mw(通过gpc-dri测量)是5000-1000000g/mol,供选择地200000-1000000g/mol,供选择地250000-800000g/mol,供选择地300000-600000g/mol,供选择地300000-500000g/mol。
通常,本文所生产的聚合物的mw/mn(通过gpc-dri测量)大于1-40,优选1-20,优选1.1-15,优选1.2-10,优选1.3-5,优选1.4-4。
通常,本文所生产的聚合物(通常地丙烯-乙烯共聚物)的mw(通过gpc-dri测量)是5000-1000000g/mol(优选200000-750000g/mol,优选250000-500000g/mol,优选250000-300000g/mol,优选250000-350000g/mol),和/或mw/mn大于1-40(供选择地1.1-20,供选择地1.2-10,供选择地1.3-5,1.4-4,供选择地1.4-3)。
在一种优选的实施方案中,本文所生产的聚合物具有单峰或者多峰分子量分布,其是通过凝胶渗透色谱法(gpc)测定的。用“单峰”表示gpc迹线具有一个峰或者拐点。用“多峰”表示gpc迹线具有至少两个峰或者拐点。拐点是这样的点,此处该曲线的二阶导数符号变化(例如从负到正或者反之亦然)。
本文所生产的聚合物的熔点(tm,dsc峰二次熔融)可以是至少145℃,或者至少150℃,或者至少152℃,或者至少153℃,或者至少154℃。例如,该聚合物的熔点可以是至少145℃-大约175℃,大约150℃-大约165℃,大约152℃-大约160℃。
本文所生产的聚合物的1%正割挠曲模量可以从大约1100mpa,大约1200mpa,大约1250mpa,大约1300mpa,大约1400mpa,或者大约1500mpa的低点到大约1800mpa,大约2100mpa,大约2600mpa,或者大约3000mpa的高点,其是根据astmd790(a,1.0mm/min)测量的。例如,该聚合物的挠曲模量可以是大约1100mpa-大约2200mpa,大约1200mpa-大约2000mpa,大约1400mpa-大约2000mpa,或者大约1500mpa或者更大,其是根据astmd790(a,1.0mm/min)测量的。
本文所生产的聚合物的熔体流动速率(mfr,astm1238,2.16kg,230℃)可以从大约0.1dg/min,大约0.2dg/min,大约0.5dg/min,大约1dg/min,大约15dg/min,大约30dg/min,或者大约45dg/min的低点到大约75dg/min,大约100dg/min,大约200dg/min,或者大约300dg/min的高点。
本文所生产的聚合物的支化指数(g'vis)可以是0.95或者更小,优选0.93或者更小,优选0.90或者更小,优选0.88或者更小。
令人感兴趣地,本文所生产的聚合物在较高和较低共聚单体引入率二者时都具有高的mw。例如,参见图3,其中催化剂a,b和c表现出具有较低乙烯含量的聚合物的mw接近于具有较高乙烯含量的使用相同催化剂所制造的共聚物的mw。
在本发明的一种实施方案中,本文所生产的聚合物是丙烯乙烯共聚物,其的乙烯含量是10-小于40wt%mol或者wt%)和其的mw≥0.9(供选择地1.0,供选择地1.1,供选择地1.25,供选择地1.5)倍的在相同聚合条件下(除了乙烯单体浓度)使用相同催化剂体系所生产的具有40wt%乙烯的丙烯乙烯共聚物的mw,并且两种mw是200000g/mol(gpc-dri)或者更大,优选250000g/mol或者更大,优选300000g/mol或者更大,优选350000g/mol或者更大。
有利地,所生产的聚合物是丙烯乙烯共聚物,其的mw≥0.9倍(供选择地1.0倍)的在相同聚合条件下(除了乙烯和丙烯单体浓度)使用相同催化剂体系所生产的具有40wt%乙烯的丙烯乙烯共聚物的mw,优选所述mw是200000g/mol(gpc-dri)或者更大。
在本发明的一种实施方案中,本文在聚合温度80℃或者更大(优选90℃或者更大,优选95℃或者更大,优选100℃或者更大)所生产的任何聚合物的熔点是x℃或者更大(供选择地x℃+1℃或者更大,供选择地x℃+2℃或者更大,供选择地x℃+3℃或者更大,供选择地x℃+4℃或者更大,供选择地x℃+5℃或者更大,供选择地x℃+6℃或者更大,供选择地x℃+7℃或者更大),其中x=(在相同条件下,除了聚合温度是70℃所制造的聚合物的tm)减去10℃。例如,如果在70℃所制造的聚合物的tm是155℃,则在80℃或者更高温度所制造的聚合物的tm是145℃或者更大。
共混物
在另一实施方案中,本文所生产的聚合物(优选聚乙烯或者聚丙烯)是在形成膜、模制件或者其他制品之前与一种或多种另外的聚合物组合的。其他有用的聚合物包括聚乙烯,全同立构聚丙烯,高度全同立构聚丙烯,间同立构聚丙烯,丙烯和乙烯、和/或丁烯、和/或己烯的无规共聚物,聚丁烯,乙烯乙酸乙烯酯,ldpe,lldpe,hdpe,乙烯乙酸乙烯酯,乙烯丙烯酸甲酯,丙烯酸共聚物,聚甲基丙烯酸甲酯或者任何其他可以通过高压自由基方法聚合的聚合物,聚氯乙烯,聚丁烯-1,全同立构聚丁烯,abs树脂,乙烯-丙烯橡胶(epr),硫化的epr,epdm,嵌段共聚物,苯乙烯类嵌段共聚物,聚酰胺,聚碳酸酯,pet树脂,交联聚乙烯,乙烯和乙烯醇(evoh)的共聚物,芳族单体的聚合物例如聚苯乙烯,聚-1酯,聚缩醛,聚偏二氟乙烯,聚乙二醇和/或聚异丁烯。
在一种优选的实施方案中,所述聚合物(优选聚乙烯或者聚丙烯)在上述共混物中的存在量是10-99wt%,基于共混物中聚合物的重量,优选20-95wt%,甚至更优选至少30-90wt%,甚至更优选至少40-90wt%,甚至更优选至少50-90wt%,甚至更优选至少60-90wt%,甚至更优选至少70-90wt%。
上述共混物可以如下来生产:将本发明的聚合物与一种或多种聚合物(如上所述)混合,将反应器串联在一起来制造反应器共混物或者在同一反应器中使用大于一种催化剂来生产多种聚合物物质。该聚合物可以在置于挤出机之前混合在一起或者可以在挤出机中混合。
所述共混物可以使用常规装置和方法形成,例如干混各个组分和随后在混合器中熔融混合,或者将所述组分直接在混合器例如banbury混合器,haake混合器,brabender密炼机,或者单螺杆或者双螺杆挤出机中混合,其可以包括配混挤出机和侧臂挤出机,其直接用于聚合方法下游,其可以包括将树脂粉末或者粒料在膜挤出机的料斗中共混。此外,根据需要,添加剂可以包括在共混物中,共混物的一种或多种组分中,和/或共混物形成的产物中,例如膜中。这样的添加剂是本领域公知的,并且可以包括例如:填料;抗氧化剂(例如受阻酚例如irganoxtm1010或者irganoxtm1076,可获自ciba-geigy);亚磷酸酯(例如irgafostm168,可获自ciba-geigy);抗粘着(anti-cling)添加剂;增粘剂例如聚丁烯,萜烯树脂,脂肪族和芳族烃树脂,碱金属和甘油硬脂酸酯,和氢化松香;uv稳定剂;热稳定剂;抗粘连剂;脱模剂;抗静电剂;颜料;着色剂;染料;蜡;二氧化硅;填料;滑石;等等。
膜
具体地,任何前述聚合物,例如前述聚丙烯或者其共混物,可以用于各种终端用途应用。这样的应用包括例如单层或者多层吹塑,挤出和/或收缩膜。这些膜可以通过任何数量的公知的挤出或者共挤出技术来制造,例如吹塑膜泡膜加工技术,其中该组合物可以在熔融态挤出通过环形模口,然后膨胀来形成单轴或者双轴取向熔体,然后冷却来形成管状吹塑膜,其然后可以轴向切开和展开来形成平膜。膜可以随后不取向,单轴取向或者双轴取向到相同或者不同的程度。所述膜的一个或多个层可以以横向和/或纵向来取向到相同或者不同的程度。单轴取向可以使用通常的冷拉伸或者热拉伸方法来完成。双轴取向可以使用拉幅机装置或者双膜泡方法来完成,并且可以在各个层置于一起之前或之后进行。例如,聚乙烯层可以挤出涂覆或者层合到取向的聚丙烯层上,或者聚乙烯和聚丙烯可以一起共挤出成膜,然后取向。同样,取向的聚丙烯可以层合到取向的聚乙烯上或者取向的聚乙烯可以涂覆到聚丙烯上,然后任选地该组合物可以甚至进一步取向。通常所述膜是在纵向(md)上以至多15,优选5-7的比率取向的,和在横向(td)上以至多15,优选7-9的比率取向的。然而,在另一实施方案中,所述膜在md和td二者的方向上取向到相同的程度。
所述膜的厚度可以根据目标应用而变化;然而,膜厚度1-50μm通常是合适的。打算用于包装的膜通常是10-50μm厚度。密封层的厚度通常是0.2-50μm。在膜的内表面和外表面二者上可以存在密封层或者该密封层可以仅仅存在于内表面或者外表面上。
在另一实施方案中,一个或多个层可以通过电晕处理,电子束辐照,γ辐照,火焰处理或者微波来改性。在一种优选的实施方案中,表面层之一或之二是通过电晕处理来改性的。
实验
mao是甲基铝氧烷(30wt%甲苯溶液),获自albemarle。
tonal是三正辛基铝。
me2si(2-ipr,4-3'5'二-t-buphind)(2-me,4-2ph-phind)zrcl2和me2si(2-ipr,4-3'5'二-t-buphind)(2-me,4-4-3'5'二-t-buphind)zrcl2是如us2015/0025208所述生产的。
mcn1是二甲基甲硅烷基(4-oph.2.-2-正己基-茚基)(2-甲基-4-(3',5'-二叔丁基-4'-甲氧基-苯基)-茚基)二氯化锆。
mcn2是二甲基甲硅烷基双(4-oph.2.-2-正己基-茚基)二氯化锆。
mcn3是二甲基甲硅烷基双(4-oph.2.-2正丁基-茚基)二氯化锆。
mcn4是二甲基甲硅烷基(4-oph.2.-2正丁基-茚基)(2-甲基-4-(3',5'-二叔丁基-4'-甲氧基-苯基)-引达省基)二氯化锆。
mcn5是二甲基甲硅烷基(4-oph.2.-2-c-丙基-茚基)(2-甲基-4-(3',5'-二叔丁基-4'-甲氧基-苯基)-茚基)二氯化锆。
mnc6是二甲基甲硅烷基(4-oph.2.-2-c-丙基-茚基)(2-甲基-4-(3',5'-二叔丁基-苯基)-茚基)二氯化锆。
mcn7是二甲基甲硅烷基(4-oph.2.-2-甲基-茚基)(2-异丙基-4-(3',5'-二叔丁基-4'-甲氧基-苯基)-茚基)二氯化锆。
mcn8是二甲基甲硅烷基双(2-c-丙基-4-(3',5'-二叔丁基-苯基)-茚基)二氯化锆。
mcn9是二甲基甲硅烷基双(2-甲基-4-(3',5'-二叔丁基-4'-甲氧基-苯基)-茚基)二氯化锆。
mcn10是二甲基甲硅烷基(4(4'叔丁基-苯基)-2-甲基-茚基)(2-异丙基-4-(4'叔丁基-苯基)-茚基)二甲基锆。
mcn11是二甲基甲硅烷基(4-苯基-2-甲基-引达省基)(2-异丙基-4-(4叔丁基-苯基)-茚基)二甲基锆。
mcn12是二甲基甲硅烷基双(4-苯基-2-正丁基-茚基)二氯化锆(6:1外消旋:内消旋之比)。
mcn13是二甲基甲硅烷基双(4-苯基-2-正丁基-茚基)二氯化锆(1:2外消旋:内消旋之比)。
mcn14是二甲基甲硅烷基(4-oph.2.-2-正己基-茚基)(2-甲基-4-(3',5'-二叔丁基-4'-甲氧基-苯基)-茚基)二甲基锆。
催化剂a是负载于smao上的mcn1。
催化剂b是负载于smao上的mcn2。
催化剂c是负载于smao上的mcn3。
催化剂d(对比)是负载于smao上的mcn8。
催化剂e(对比)是负载于smao上的mcn10。
催化剂f(对比)是负载于smao上的mcn9。
茂金属合成:
mcn1
4-([1,1'-联苯基]-2-基)-2-nhex-1h-茚:将化合物4-([1,1'-联苯基]-2-基)-2-溴-1h-茚(15g,43.2mmol,1当量)和无水甲苯(150ml)的溶液用双(三苯基膦)二氯化钯(ii)(3.5g,4.3mmol,0.1当量)处理。在搅拌10分钟后,逐滴加入2m己基溴化镁的二乙醚溶液(112ml,224.6mmol,5.2当量)。该反应在60℃加热5小时。将该反应用冰浴冷却,用1nhcl酸化到ph3和用乙酸乙酯(3x500ml)萃取。将合并的有机层用饱和盐水(800ml)洗涤,在硫酸钠上干燥和减压下浓缩。将残留物在硅胶(200g)上纯化,用庚烷洗提来产生作为浅黄色油的化合物7(7g,46%收率)。
{1-[4-(3’,5’-二叔4’-甲氧基丁基苯基)-2-甲基茚化锂]}:将4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基-茚(15.0g,43.1mmol)在二乙醚(200ml)中的预冷却溶液用nbuli(2.5m己烷溶液,18.1ml,45.3mmol)处理。将该反应在室温搅拌15h。然后将全部挥发物蒸发。将残留物用戊烷(10ml)洗涤和真空干燥来产生白色固体(15.15g)。
氯二甲基[4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-茚基]硅烷:将1-[4-(3,5-二叔4-甲氧基丁基苯基)-2-甲基茚化锂](15.1g,42.8mmol)在二乙醚(100ml)中的预冷却溶液用me2sicl2(27.4g,214.0mmol)处理,并且将该白色淤浆在室温搅拌5h。将全部挥发物在减压下蒸发。将该残留物用己烷(100mlx2)萃取,和将合并的滤出液真空下浓缩干燥来产生白色泡沫(18.36g)。
二甲基甲硅烷基[4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-茚基]三氟甲烷磺酸酯:将氯二甲基[4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基-茚基]硅烷(18.34g,41.7mmol)在甲苯(100ml)中的溶液用三氟甲烷磺酸银(11.2g,43.8mmol)在搅拌的同时进行处理。将该白色淤浆在室温搅拌5h。真空下除去甲苯和将残留物用己烷(100mlx2)萃取。将收集的滤出液真空下浓缩来产生作为产物的无色泡沫(22.82g)。
[1-(4-oph.2.)-2-己基-茚化锂]:将4-oph.2.-2-己基-茚(15.0g,42.6mmol)在二乙醚(100ml)中的预冷却溶液用nbuli(2.5m己烷溶液,17.9ml,44.7mmol)处理。将该反应在室温搅拌3h。然后将全部挥发物蒸发。将残留物用己烷(20mlx2)洗涤和真空下干燥来产生作为产物的白色固体(14.21g)。
(4-oph.2.-2-己基-茚基)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-茚基)二甲基硅烷:将二甲基甲硅烷基[4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基-茚基]三氟甲烷磺酸酯(22.73g,39.2mmol)在二乙醚(100ml)中的预冷却溶液用[1-(4-oph.2.-2-己基茚化锂)](14.03g,39.2mmol)处理。将该溶液在室温搅拌过夜。蒸发二乙醚。将残留物通过快速色谱法(硅胶,洗提剂:己烷)纯化来产生淡黄色油(13.24g)。
二锂二甲基甲硅烷基(4-oph.2.-2-己基茚化物)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基茚化物):将nbuli(2.5m,14.3ml,35.79mmol)加入上述产物(13.20g,17.46mmol)在二乙醚(100ml)中的预冷却溶液中。将该溶液在室温搅拌3h。将全部挥发物在真空下除去。将残留物用戊烷(15mlx2)洗涤和真空干燥来产生所述二锂化合物(12.11g)。
二甲基甲硅烷基(4-oph.2.-2-己基茚基)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基茚基)二氯化锆:将二锂二甲基甲硅烷基(4-oph.2.-2-环丙基茚化物)(4-(3,5-二叔丁基苯基)-2-甲基茚化物(12.06g,15.7mmol)在甲苯(100ml)中的预冷却溶液用zrcl4(3.79g,1.17mmol)处理。将该混合物在室温搅拌过夜。将该混合物通过celite过滤来除去licl和蒸发干燥。将残留物用己烷(50ml)洗涤来获得作为两种异构体混合物的固体。该混合物在甲苯(20ml,100℃-40℃)重结晶来产生相应的内消旋异构体茂金属(361mg,外消旋/内消旋之比=1:22)。将合并的滤出液浓缩和重结晶(10ml甲苯和5ml己烷,回流到室温)来提供外消旋/内消旋之比=15:1的混合物。该混合物进一步重结晶(10ml甲苯和6ml己烷,回流到室温)来获得外消旋异构体(623mg,外消旋/内消旋之比=22:1)。1hnmr(400mhz,c6d6,23℃),外消旋形式异构体:δ8.26(dd,1h),7.91(s,2h),7.51(d,1h),7.43(dd,1h),7.36-7.32(m,1h),7.29(d,1h),7.25(td,1h),7.18-7.09(m,5h),6.95-6.83(m,5h),6.69(dd,1h),3.41(s,3h),2.76-2.66(m,1h),2.48-2.38(m,1h),1.96(s,3h),1.57(s,18h),1.47-1.13(m,8h),0.93-0.87(m,6h),0.65(s,3h);内消旋形式异构体:1hnmr(400mhz,c6d6,23℃)δ8.22-8.18(m,1h),7.90(s,2h),7.38(dd,2h),7.31-7.28(m,2h),7.19-7.09(m,2h),7.05-7.71(m,3h),6.96-6.78(m,4h),6.75(dd,1h),6.67(s,1h),6.58(dd,1h),3.39(s,3h),2.81-2.71(m,1h),2.66-2.56(m,1h),2.18(s,3h),1.54(s,18h),1.40-1.12(m,8h),0.91(t,3h),0.81(s,3h),0.76(s,3h)。
mcn2
{1-[(4-oph.2.-2-n己基)茚化锂]}:将nbuli(2.5m,8.2ml,20.5mmol)加入到搅拌的4-([1,1'-联苯基]-2-基)-2-nhex-1h-茚(6.55g,18.58mmol)在二乙醚(100ml)中的预冷却溶液中。将该溶液室温搅拌19小时。蒸发全部挥发物。将残留物真空干燥来产生含有0.08当量的et2o的粗产物(6.07g)。该产物无需进一步纯化而使用。
氯二甲基[4-oph.2.-2-n己基-茚基]硅烷:将me2sicl2(10g,77.48mmol)加入到上述锂盐(1.97g,5.40mmol)在二乙醚(60ml)中的预冷却溶液中。加入另外的二乙醚(10ml)。将该白色淤浆在室温搅拌17h。真空除去全部挥发物。将该残留物用己烷萃取(50ml一次,10ml一次)和将滤出液真空下浓缩来产生所述产物(2.19g)。该产物无需进一步纯化而使用。
二甲基甲硅烷基[4-oph.2.-2-n己基-茚基]三氟甲烷磺酸酯:将三氟甲烷磺酸银(1.31g,5.098mmol)加入搅拌的上述产物(2.16g,4.853mmol)在甲苯(25ml)中的溶液中。加入另外的甲苯(10ml)。将该淤浆在室温搅拌1h。真空下除去甲苯和将残留物用己烷萃取(40ml一次,10ml一次)。将己烷滤出液真空下浓缩来产生所述产物(2.55g)。该产物无需进一步纯化而使用。
双(4-oph.2.-2-n己基-茚基)二甲基硅烷:将{1-[1-[(4-oph.2.-2-n己基)茚化锂]}(et2o)0.08(1.62g,4.446mmol)加入二甲基甲硅烷基[4-oph.2.-2-n己基-茚基]三氟甲烷磺酸酯(2.48g,4.439mmol)在二乙醚(40ml)中的预冷却溶液中。加入另外的二乙醚(10ml)。将该反应在室温搅拌19h。蒸发全部挥发物。将该残留物用己烷萃取(50ml一次,10ml一次)和将该滤出液真空下浓缩来产生粗产物(3.28g)。该产物无需进一步纯化而使用。
二甲基甲硅烷基双(4-oph.2.-2-n己基-茚化二锂):将nbuli(2.5m,3.5ml,8.75mmol)加入上述粗产物(3.22g)在二乙醚(30ml)和己烷(15ml)中的预冷却溶液中。将该溶液室温搅拌24h。真空下除去全部挥发物。将残留物用己烷洗涤(20ml两次)和真空干燥来产生含有0.54当量et2o的粗产物(3.29g)。
二甲基甲硅烷基双(4-oph.2.-2-n己基-茚基)二氯化锆:将zrcl4(0.96g,4.119mmol)加入上述粗产物(3.27g)在甲苯(40ml)中的预冷却溶液中。加入另外的甲苯(10ml)。将该混合物在室温搅拌18h。真空下除去全部挥发物。将残留物用己烷萃取(60ml一次,10ml一次)。然后将己烷不溶物萃取到甲苯中(40ml一次,10ml一次)。将甲苯滤出液真空下浓缩干燥来产生粗产物,其是1/1.2比率的外消旋/内消旋混合物(1.15g)。加入甲苯(4ml)和己烷(32ml)。将该淤浆加热到回流和然后冷却回室温。该混合物在室温搅拌3天。将沉淀物分离,用己烷洗涤(5ml两次)和真空干燥来产生外消旋/内消旋之比是1/1.6(0.99g)的固体。从二乙醚中的另外的多个分步结晶提供了外消旋/内消旋之比是大约50/1的粗产物(0.28g)加上一些不溶杂质。向这种粗产物中加入ch2cl2(18ml)。将该混合物过滤和将不溶物用另外的ch2cl2洗涤(18ml一次,5ml一次)。将滤出液和洗涤物合并和蒸发干燥。所获得的固体用二乙醚(5ml)洗涤和真空干燥来产生产物(0.15g,外消旋/内消旋=40/1)。1hnmr(400mhz,cd2cl2,23℃):外消旋:δ7.64(m,2h),7.49(m,2h),7.40-7.46(m,6h),7.11(m,2h),7.04-7.08(m,10h),6.91(m,2h),6.32(s,2h),2.54(m,2h),2.10(m,2h),1.32-1.08(m,22h),0.88(t,6h)。
mcn3
[1-(4-oph.2.)-2-丁基-茚化锂]:将4-oph.2.-2-丁基-茚(7.80g,24.1mmol)在二乙醚(50ml)中的预冷却溶液用nbuli(2.5m己烷溶液,10.1ml,25.3mmol)处理。将该反应室温搅拌3h。然后蒸发全部挥发物。将残留物用己烷(10mlx2)洗涤和真空干燥来产生灰白色固体作为产物(7.09g)。
氯二甲基[(4-oph.2.)-2-丁基-茚基]硅烷:将[1-(4-oph.2.)-2-丁基-茚化锂](3.30g,10.0mmol)在二乙醚(50ml)中的预冷却溶液用me2sicl2(6.50g,50.0mmol)处理,和将所形成的白色淤浆在室温搅拌过夜。将全部挥发物减压下蒸发。将该残留物用己烷(30mlx2)萃取,和将合并的滤出液真空下浓缩干燥来产生无色油(2.94g)。
二甲基甲硅烷基(4-oph.2.-2-丁基-茚基)三氟甲烷磺酸酯:将氯二甲基(4-oph.2.-2-丁基-茚基)硅烷(2.90g,6.97mmol)在甲苯(30ml)中的溶液用三氟甲烷磺酸银(1.96g,7.67mmol)在搅拌的同时处理。将该白色淤浆室温搅拌5h。真空下蒸发甲苯和将残留物用己烷(30mlx2)萃取。将滤出液真空下浓缩来产生无色油作为产物(3.60g)。
双(4-oph.2.-2-丁基-茚基)二甲基硅烷:将二甲基甲硅烷基(4-oph.2.-2-丁基-茚基)三氟甲烷磺酸酯(3.50g,6.60mmol)在二乙醚(30ml)中的预冷却溶液用[1-(4-oph.2.)-2-丁基-茚化锂](2.18g,6.60mmol)处理。将该溶液室温搅拌3小时。蒸发二乙醚。将残留物用溶剂(与30ml甲苯和30ml己烷混合)萃取。将合并的滤出液浓缩和在真空进一步干燥来获得灰白色固体作为产物(3.24g)。
二甲基甲硅烷基双(4-oph.2.-2-丁基-茚化二锂):将nbuli(2.5m,3.7ml,9.26mmol)加入双(4-oph.2.-2-丁基-茚基)二甲基硅烷(3.18g,4.52mmol)在二乙醚(30ml)中的预冷却溶液中。将该溶液室温搅拌3h。真空下除去全部挥发物。将残留物用己烷洗涤(10mlx2)和真空干燥来产生淡黄色泡沫(2.45g)。
二甲基甲硅烷基双(4-oph.2.-2-丁基-茚基)二氯化锆:将二甲基甲硅烷基双(4-oph.2.-2-丁基-茚化二锂)(2.37g,3.31mmol)在甲苯(30ml)中的预冷却溶液用zrcl4(0.76g,3.31mmol)处理。将该混合物室温搅拌过夜。将该混合物蒸发干燥。将残留物用热环己烷(50ml)萃取。将合并的滤出液减压浓缩和用己烷(20ml)洗涤来获得橙色固体,其是两种异构体的混合物。该混合物重结晶(2ml甲苯和18ml己烷,回流到室温)来提供混合物(520mg,湿,外消旋/内消旋之比=10:1)。然后将该混合物进一步重结晶(1.5ml甲苯和13.5ml己烷,回流到室温)来提供外消旋异构体(100mg,外消旋/内消旋之比=68:1)。1hnmr(400mhz,c6d6,23℃),外消旋形异构体:δ8.26-8.23(m,2h),7.39(s,1h),7.36(s,1h),7.35(d,1h),7.33(d,1h),7.22-7.12(m,4h),7.10-7.05(m,6h),6.90-6.78(m,8h),6.70(dd,2h),2.66-2.55(m,2h),2.41-2.30(m,2h),1.29-1.19(m,4h),1.13-1.02(m,4h),0.81(t,6h),0.75(s,6h)。
mcn4
氯二甲基(4-oph.2.-2-丁基-茚-1-基)硅烷:将[1-(4-oph.2.)-2-丁基-茚化锂](3.30g,10.0mmol)在二乙醚(50ml)中的预冷却溶液用me2sicl2(6.45g,50.0mmol)处理,和将所形成的白色淤浆在室温搅拌过夜。蒸发全部挥发物。将残留物用己烷(20mlx2)萃取,和将合并的滤出液减压浓缩来获得无色油(3.91g)。
二甲基甲硅烷基(4-oph.2.-2-丁基-茚-1-基)三氟甲烷磺酸酯:将氯二甲基(4-oph.2.-2-丁基-茚-1-基)硅烷(3.90g,9.4mmol)在甲苯(30ml)中的预冷却溶液用三氟甲烷磺酸银(2.64g,10.3mmol)在搅拌的同时进行处理。将该白色淤浆室温搅拌3小时。减压下除去甲苯,和将残留物用己烷(20mlx2)萃取。将收集的滤出液减压下过滤来获得作为产物的无色油(4.88g)。
{4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省化锂}:将8-(3,5-二叔丁基-4-甲氧基苯基)-6-甲基-1,2,3,5-四氢对称引达省(3.88g,10.0mmol)在二乙醚(20ml)中的预冷却溶液用nbuli(2.5m己烷溶液,4.2ml,10.5mmol)处理。将该反应在室温搅拌过夜。然后蒸发全部挥发物。将残留物用己烷洗涤(20mlx2)和真空下干燥来产生橙色固体(3.60g)。
(4-oph.2.-2-丁基-茚基)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省基)二甲基硅烷:将二甲基甲硅烷基(4-oph.2.-2-丁基-茚-1-基)三氟甲烷磺酸酯(4.80g,9.06mmol)在二乙醚(30ml)的预冷却溶液用1-[4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省化锂]固体(3.57g,9.06mmol)处理。将该溶液在室温搅拌过夜。将二乙醚蒸发。将残留物用己烷萃取(30mlx2)。将合并的滤出液浓缩干燥和在真空干燥来获得无色泡沫(6.70g)。
二锂二甲基甲硅烷基(4-oph.2.-2-丁基茚化物)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省化物):将nbuli(2.5m,7.1ml,17.83mmol)加入(4-oph.2.-2-丁基-茚基)(4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省基)二甲基硅烷(6.68g,8.70mmol)在二乙醚(50ml)中的预冷却溶液中。将该混合物室温搅拌3小时。减压下除去全部挥发物。将残留物用冷己烷(30ml)洗涤和真空下干燥来产生橙色固体(6.247g)。
二甲基甲硅烷基(4-oph.2.-2-丁基茚基)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省基)二氯化锆:将二锂二甲基甲硅烷基(4-oph.2.-2-丁基茚化物)(4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基-1,5,6,7-四氢对称引达省化物)(6.20g,7.95mmol)在甲苯(50ml)中的预冷却溶液用粉末zrcl4(1.83g,7.95mmol)预处理。将该混合物室温搅拌5小时。然后将该混合物减压下浓缩,和将该残留物用溶剂(与25ml甲苯和20ml己烷混合)萃取。将合并的滤出液浓缩。将所形成的残留物重结晶(10ml甲苯和50ml己烷,回流到室温)。然后将所收集的固体进一步重结晶(30ml甲苯,回流到室温)来获得内消旋-异构体(853mg,外消旋/内消旋之比<1:100)。将来自于第一重结晶的滤出液浓缩和将残留物重结晶(10ml甲苯和50ml己烷,回流到室温)来提供外消旋异构体(351mg,外消旋/内消旋之比=39:1)。1hnmr(400mhz,c6d6,23℃),内消旋形式异构体:δ8.23-8.17(m,1h),7.83(bs,1h),7.52(d,1h),7.38(s,1h),7.32-7.28(m,1h),7.22-7.10(m,3h),7.07-7.03(m,2h),6.97(dd,1h),6.88-6.77(m,4h),6.63-6.57(m,2h),3.45(s,3h),3.08-2.55(m,6h),2.16(s,3h),1.85-1.65(m,2h),1.55(s,18h),1.45-1.16(m,2h),1.16-1.08(m,2h),0.91(s,3h),0.85(t,3h),0.76(s,3h);外消旋形式异构体:δ8.28(dd,1h),7.84(bs,1h),7.46(s,1h),7.37-7.23(m,3h),7.19-7.08(m,xh),6.97(s,1h),6.94-6.83(m,4h),6.68(dd,1h),3.47(s,3h),3.12-3.02(m,1h),2.98-2.72(m,4h),2.50-2.40(m,1h),1.95(s,3h),1.84-1.72(m,2h),1.59(s,18h),3.08-2.55(m,6h),2.16(s,3h),1.85-1.65(m,2h),1.55(s,18h),1.38-1.10(m,4h),0.94(s,3h),0.87(t,3h),0.69(s,3h)。
mcn12和mnc13
4-苯基-1h-茚:将250ml烧瓶加入4-溴-1h-茚(10.00g,51.55mmol),苯基硼酸(6.60g,54.12mmol),碳酸钾(10.67g,77.33mmol),四丁基溴化铵(3.42g,10.31mmol),双(三苯基膦)二氯化钯(ii)(1.80g,2.50mmol),水(150ml)和乙醇(15ml)。将该反应在n2气氛下回流3小时。将该反应冷却和用己烷(3x100ml)萃取。将合并的有机层在na2so4上干燥和减压下浓缩。将所形成的残留物通过快速色谱法在硅胶(洗提剂:己烷)上纯化来获得作为无色油的产物(8.21g)。
2-溴-7-苯基-2,3-二氢-1h-茚-1-醇:将4-苯基-1h-茚(8.10g,42.2mmol)在二甲基亚砜(50ml)和水(1ml)中的预冷却(5℃)溶液用n-溴琥珀酰亚胺(8.26g,46.4mmol)一次性处理,和使得该反应自然温热到室温和搅拌3小时。将该混合物倾倒入水(500ml)中和用甲苯(3x50ml)萃取。将该合并的有机相用水(100ml)洗涤和在na2so4上干燥。将甲苯中的粗产物无需进一步纯化而用于接下来的步骤。
2-溴-4-苯基-1h-茚:将来自于前述步骤的溶液用对甲苯磺酸一水合物(0.4g,6.3mmol)处理和将该混合物回流6小时,同时用dean-stark阱除水。将该混合物冷却和减压下浓缩。将该残留物通过快速色谱法在硅胶(洗提剂:己烷)上纯化来获得作为白色固体的2-溴-4-苯基-1h-茚(7.36g)。
2-丁基-4-苯基-1h-茚:在手套箱中,将正丁基氯化镁(15.0ml,2.0m的thf溶液,29.54mmol)加入2-溴-4-苯基-1h-茚(7.25g,26.85mmol)和pdcl2(dppf)·dcm(1.10g,1.34mmol)在30ml的thf中的溶液中。将该反应加热高到45℃和在这个温度搅拌3小时。将该反应从手套箱移出和用200ml水淬冷。将该混合物用己烷(50mlx2)萃取。将该合并的有机相在na2so4上干燥和减压浓缩。将该残留物通过硅胶色谱法纯化(洗提剂:己烷)来获得作为无色油的产物(2.93g)。
[1-(4-苯基-2-丁基茚化锂)]:将2-丁基-4-苯基-1h-茚(2.71g,10.9mmol)在二乙醚(20ml)中的预冷却溶液用nbuli(2.5m己烷溶液,5.7ml,11.4mmol)处理。将该反应在室温搅拌3h。然后蒸发全部的挥发物。将该残留物用己烷(20ml)洗涤和真空下干燥来产生作为产物的绿色-黄色固体(2.700g)。
氯二甲基(4-苯基-2-丁基-茚基)硅烷:将[1-(4-苯基-2-丁基茚化锂)](1.35g,5.31mmol)在二乙醚(20ml)中的预冷却溶液用me2sicl2(3.43g,26.57mmol)处理,和将所形成的白色淤浆在室温搅拌过夜。在19小时搅拌后,减压下蒸发全部挥发物。将残留物用己烷(20mlx2)萃取,和将合并的滤出液真空下浓缩干燥来产生作为产物的无色油(1.79g)。
二甲基甲硅烷基(4-苯基-2-丁基-茚基)三氟甲烷磺酸酯:将氯二甲基(4-苯基-2-丁基-茚基)硅烷(1.78g,5.26mmol)在甲苯(20ml)中的溶液用三氟甲烷磺酸银(1.48g,5.78mmol)在搅拌同时处理。将该白色淤浆在室温搅拌3h。将甲苯真空下蒸发和将残留物用己烷(10mlx2)萃取。将该滤出液真空浓缩来产生作为产物的无色油(2.17g)。
双(4-苯基-2-丁基-茚基)二甲基硅烷:将二甲基甲硅烷基(4-苯基-2-丁基-茚基)三氟甲烷磺酸酯(2.10g,4.62mmol)在二乙醚(20ml)中的预冷却溶液用[1-(4-苯基-2-丁基茚化锂)](1.17g,4.62mmol)处理。将该溶液室温搅拌5小时。蒸发二乙醚。将残留物用己烷(2x10ml)萃取。将合并的滤出液浓缩和真空进一步干燥来获得作为产物的灰白色泡沫(2.56g)。
二甲基甲硅烷基双(4-苯基-2-丁基茚化二锂):将nbuli(2.5m己烷溶液,3.7ml,9.28mmol)加入到双(4-苯基-2-丁基-茚基)二甲基硅烷(2.50g,4.53mmol)在二乙醚(20ml)中的预冷却溶液中。将该溶液室温搅拌3h。真空下除去全部挥发物。将残留物用己烷(10mlx2)洗涤和真空干燥来产生作为期望的二锂盐的浅粉色固体(2.38g)。
二甲基甲硅烷基双(4-苯基-2-丁基-茚基)二氯化锆:将二甲基甲硅烷基双(4-苯基-2-丁基茚化二锂)(2.30g,4.08mmol)在甲苯(30ml)中的预冷却溶液用zrcl4(0.938g,4.08mmol)处理。将该混合物室温搅拌整个周末。在64小时搅拌后,将该混合物浓缩干燥。将残留物用甲苯(30mlx2)萃取。将合并的滤出液减压浓缩和用己烷(20ml)洗涤来获得橙色固体,其是两种异构体的混合物。该混合物是重结晶(20ml二氯甲烷和10ml二乙醚,回流到室温,老化16小时)。将该淤浆过滤和将所述薄饼用己烷(10ml)洗涤来提供外消旋异构体(黄色,433mg,外消旋/内消旋之比=6.7:1,mnc12)。将该滤出液减压浓缩和将残留物用二乙醚(30ml)洗涤来获得富含内消旋异构体的混合物(橙色,651mg,外消旋/内消旋之比=1:1.8,mcn13)。1hnmr(400mhz,c6d6,23℃),外消旋形式异构体:δ7.88(dd,4h),7.57(d,2h),7.30(dd,2h),7.27(s,2h),7.21(t,4h),7.10-7.04(m,2h),6.90(dd,2h),2.76-2.66(m,2h),2.50-2.40(m,2h),1.36-1.22(m,4h),1.19-1.10(m,4h),0.96(s,6h),0.75(t,6h);内消旋形式异构体(从外消旋/内消旋=1:1.8混合物确定):δ7.83(dd,4h),7.55(d,2h),7.21(dd,6h),7.13-6.99(m,4h),6.75(dd,2h),2.85-2.70(m,4h),1.40-1.05(m,8h),1.10(s,3h),0.96(s,3h),0.75(t,6h)。
mcn14
二甲基甲硅烷基(4-oph.2.-2-己基茚基)(4-(3’,5’-二叔丁基-4’-甲氧基苯基)-2-甲基茚基)二甲基锆:将外消旋-二甲基甲硅烷基(4-oph.2.-2-己基茚基)(4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基茚基)二氯化锆(mcn1)(具有0.5当量甲苯)(1.1g)在二乙醚(100ml)中的溶液在-35℃预冷却15min。加入memgbr(3.5ml的3m二乙醚溶液)和将该反应在室温搅拌70h。蒸发全部的挥发物。将该残留物用己烷萃取(60ml一次,20ml三次)。将滤出液合并和蒸发干燥。加入己烷(15ml)和将该淤浆在-35℃放置1天来产生0.35g的二甲基甲硅烷基(4-oph.2.-2-己基茚基)(4-(3,5-二叔丁基-4-甲氧基苯基)-2-甲基茚基)二甲基锆,外消旋/内消旋:~44/1(mcn14)。1hnmr(400mhz,c6d6,23℃),外消旋异构体:δ7.85(m,3h),7.52(d,1h),7.42-7.40(m,2h),7.31(d,1h),7.25-7.22(m,3h),7.12-7.18(m,3h),6.99-6.88(m,4h),6.77(s,1h),6.72(dd,1h),3.41(s,3h),2.65-2.59(m,1h),2.17-2.09(m,1h),1.92(s,3h),1.52(s,18h),1.45-1.18(m,8h),0.94(t,3h),0.86(s,3h),0.66(s,3h),-0.54(s,3h),-0.61(s,3h)。
负载的甲基铝氧烷(smao)
smao是如下来制备的:davison948tm二氧化硅(20.8606g,在130℃煅烧)在121ml甲苯中制浆和在冰箱中冷却(-35℃)。mao(50.5542g的30%wt甲苯溶液)分3份缓慢加入,并且在添加之间将该二氧化硅淤浆返回冰箱几分钟(大约2min)。将该淤浆室温搅拌2小时,用细玻璃料过滤器过滤,在80ml甲苯中在室温重新制浆15min,然后再次过滤。将该固体在80ml甲苯中在80℃重新制浆30min,然后过滤。将该固体在80ml甲苯中在80℃重新制浆30min,然后最后一次过滤。将celstir和固体用40ml甲苯洗涤。然后将该固体用戊烷洗涤和真空干燥24小时。收集了28.9406g的自由流动的白色粉末。
制备负载的催化剂
负载的催化剂a:将mcn1(26.4mg,0.0288mmol)和mao(0.2409g的30重量%甲苯溶液)与2ml甲苯在20ml小瓶中合并在一起和搅拌1h。将smao(0.7200g)在20ml甲苯中制浆和冷却到-35℃几分钟。将该催化剂溶液加入所述淤浆中。将该淤浆搅拌1h,偶尔置于冰箱中来保持温度稍低于rt。该淤浆然后在40℃搅拌2h。将该淤浆过滤和将固体在60℃的20ml甲苯中重新制浆30min,然后再次过滤。将该固体在60℃重新制浆两次以上。该celstir然后用20ml甲苯洗出,其也用于洗涤所述固体。该固体用戊烷洗涤两次和真空干燥来产生0.6000g粉色固体。
负载的催化剂b:将mcn2(28.8mg,0.0313mmol)和mao(0.2620g的30重量%甲苯溶液)与2ml甲苯一起合并到20ml小瓶中,并且搅拌1h。将smao(0.7885g)在15ml甲苯中制浆和冷却到-35℃一分钟。将该催化剂溶液加入所述淤浆。将该淤浆搅拌1h,偶尔置于冰箱中来保持温度稍低于rt。该淤浆然后在40℃搅拌2h。将该淤浆过滤和将固体在60℃的15ml甲苯中重新制浆30min,然后再次过滤。将该固体在60℃重新制浆两次以上。该celstir然后用15ml甲苯洗出,其也用于洗涤所述固体。该固体用戊烷洗涤两次和真空干燥来产生0.7054g的粉色固体。
催化剂c,d和f是通过类似方法来制备。
负载的催化剂e(对比):将mcn10(29.3mg,0.0402mmol)和mao(0.3316g的30重量%甲苯溶液)与2ml甲苯一起合并到20ml小瓶中,并且搅拌1h。将smao(1.0079g)在15ml甲苯中制浆和冷却到-35℃几分钟。将该催化剂溶液加入所述淤浆。将该淤浆搅拌1h,偶尔置于冰箱中来保持温度稍低于rt。该淤浆然后在40℃搅拌2h。将该淤浆过滤和将固体在60℃的15ml甲苯中重新制浆30min,然后再次过滤。将该固体在60℃重新制浆两次以上。该celstir然后用15ml甲苯洗出,其也用于洗涤所述固体。该固体用戊烷洗涤两次和真空干燥来产生0.9041g的粉色固体。
用于小规模聚合的一般程序
除非另有规定,否则丙烯均聚和乙烯-丙烯共聚是在并联压力反应器中进行的,如us6306658;us6455316;wo00/09255;和murphy等人,j.am.chem.soc.,2003,125,第4306-4317页所一般描述的,其每个通过引用全部并入本文。虽然从一个反应到另一个反应,具体量,温度,溶剂,反应物,反应物比率,压力和其他变量可能需要调整,但是下面描述了在并联压力反应器中进行的通常的聚合。
对于使用未负载的茂金属催化剂的丙烯聚合和乙烯丙烯共聚,使用下面的程序:
将预称重的玻璃小瓶嵌件和一次性搅拌叶片安装到反应器的每个反应容器上,其包含48个单个反应容器。该反应器然后闭合和将丙烯气体引入每个容器来将氮气吹扫出所述系统。如果任何模块接收氢,则它在吹扫过程中加入。所述溶剂(通常是异己烷)根据设定总反应体积(包括下面的添加)接着加入到通常5ml。此时加入清除剂和/或助催化剂和/或链转移剂例如三正辛基铝的甲苯溶液(100-1000nmol)。将该容器内容物在800rpm搅拌。将丙烯作为气体加入到设定压力。将该反应器容器加热到它们的设定运行温度(通常50℃-110℃)。如果任何模块接收乙烯,则它作为气体加入到高于丙烯压力的预定压力(通常是40-220psi),同时将该反应器容器加热到设定的运行温度。
将催化剂的甲苯溶液(通常的浓度是0.2mmol/l甲苯溶液,其通常提供了大约15nmol的催化剂)注入所述反应器中。然后使该反应继续,直到该反应已经接收了预定量的压力。供选择地,可以使该反应继续设定量的时间。该反应是通过用压缩空气加压所述容器来淬灭的。在该聚合反应之后,从压力室和惰性气氛手套箱中除去含有聚合物产物和溶剂的玻璃小瓶嵌件,并且将挥发性组分是使用在高温和减压操作的genevacht-12离心机和genevacvc3000d真空蒸发器除去。然后称重该小瓶来测定聚合物产物的收率。所形成的聚合物通过快速gpc(见下面)分析来测定分子量和通过dsc(见下面)来测定熔点。
对于使用负载的茂金属催化剂的乙烯丙烯共聚,使用下面的程序:将预称重的玻璃小瓶嵌件和一次性搅拌叶片安装到反应器的每个反应容器上,其包含48个单个反应容器。该反应器然后闭合和将丙烯气体引入每个容器来将氮气吹扫出所述系统。如果任何模块接收氢,则它在吹扫过程中加入。所述溶剂(通常是异己烷)根据设定总反应体积(包括下面的添加)接着加入到通常5ml。此时加入清除剂和/或助催化剂和/或链转移剂例如三正辛基铝的甲苯溶液(100-1000nmol)。将该容器内容物在800rpm搅拌。将丙烯作为气体加入到设定压力。将该反应器容器加热到它们的设定运行温度(通常50℃-110℃)。将乙烯作为气体加入到高于丙烯压力的预定压力(通常是40-220psi),同时将该反应器容器加热到设定的运行温度。将该催化剂淤浆涡流化来将催化剂颗粒悬浮于溶液中。将缓冲甲苯(通常是100微升),催化剂的甲苯溶液(通常是3mg/ml浓度)和另一等分的甲苯(500微升)然后注入该反应器中。然后使得该反应继续直到该反应已经接收了预定量的压力。供选择地,可以使得该反应继续设定量的时间。在这个点,该反应是通过用压缩空气加压所述容器来淬灭的。在该聚合反应之后,从压力室和惰性气氛手套箱中除去含有聚合物产物和溶剂的玻璃小瓶嵌件,并且将挥发性组分是使用在高温和减压操作的genevacht-12离心机和genevacvc3000d真空蒸发器除去。然后称重该小瓶来测定聚合物产物的收率。所形成的聚合物通过快速gpc(见下面)分析来测定分子量和通过dsc(见下面)来测定熔点。数据报告在表1-4中。
为了通过gpc测定不同的分子量相关值,使用自动化的“快速gpc”系统来进行高温尺寸排阻色谱法,其通常描述在us6491816;us6491823;us6475391;us6461515;us6436292;us6406632;us6175409;us6454947;us6260407;和us6294388中;其每个就美国的目的而言通过引用完全并入本文。这个设备具有一系列的三个30cmx7.5mm线性柱,每个含有plgel10um,混合b。所述gpc系统是使用580-3390000g/mol聚苯乙烯标准物校正的。该系统是在2.0ml/min的洗提剂流速和165℃的烘箱温度进行的。将1,2,4-三氯苯用作洗提剂。将该聚合物样品溶解在1,2,4-三氯苯中,浓度是0.1-0.9mg/ml。将250ul聚合物溶液注入所述系统中。洗提剂中聚合物的浓度是使用polymercharir4检测器来监控的。所给出的分子量是相对于线性聚苯乙烯标准物的,并且是未校正的。仅仅就本发明的目的而言,该快速gpcmw(重均分子量)数据可以除以1.9来近似乙烯-丙烯共聚物的gpc-3dmw结果。同样,仅仅就本发明的目的而言,丙烯均聚物的快速gpcmw数据可以除以1.5来近似gpc-3dmw结果。
差示扫描量热法(dsc,程序-1)测量是在ta-q200仪器上进行的,来测定聚合物的熔点。样品是在220℃预先退火15分钟,然后允许冷却到室温过夜。该样品然后以速率100℃/min加热到220℃,和然后以速率50℃/min冷却。熔点是在加热期间收集的。
引入该聚合物中的乙烯的量(重量%)是通过快速ft-ir光谱法在brukervertex70ir上以反射模式测量的。样品是以薄膜形式,通过蒸发沉积技术来制备的。重量百分比乙烯获自729.8和1157.9cm-1的峰高度之比。这种方法是使用一组具有已知的wt%乙烯含量的乙烯/丙烯共聚物来校正的。
用于在连续搅拌釜反应器中的溶液聚合的一般程序(表5,实施例162-167)
聚合是在连续搅拌釜反应器系统中进行的。1l高压釜反应器装备有搅拌器,压力控制器和水冷却/蒸汽加热元件,其具有温度控制器。该反应器是在液体填充条件在超过反应混合物的鼓泡点压力的反应器压力操作的,将反应物保持在液相中。全部供料(溶剂和单体)使用pulsa供料泵泵入反应器中,并且流速是使用coriolis质量流量控制器(来自于brooks的quantim系列)来控制的,除了乙烯(其作为气体在它自身压力下流过brooks流量控制器)。将乙烯和丙烯供料合并成一个流,然后与预冷却的异己烷流(其已经冷却到至少0℃)进行混合。该混合物然后通过单个管线供给到所述反应器。在它即将进入反应器来进一步还原任何催化剂毒物之前,将清除剂溶液加入该合并的溶剂和单体流。类似地,使用isco注射器泵通过分别的管线将催化剂溶液供给到反应器。
异己烷(用作溶剂)和单体(例如乙烯和丙烯)是在氧化铝和分子筛床上纯化的。用于制备催化剂溶液的甲苯是使用相同的技术来纯化的。将三正辛基铝(tnoa)的异己烷溶液(25wt%己烷溶液,sigmaaldrich)用作清除剂溶液。催化剂mtc14是用n,n-二甲基苯胺
反应器中所生产的聚合物是通过背压控制阀离开的,该控制阀将压力降低到大气压。这引起所述溶液中未转化的单体闪蒸到气相中,其从气液分离器顶部排出。收集液体相(主要包含聚合物和溶剂)用于聚合物回收。所收集的样品首先在防护罩中空气干燥来蒸发大部分溶剂,然后在真空烘箱中在大约90℃干燥大约12小时。将该真空烘箱干燥样品称重来获得收率。转化率是基于收率和全部单体的供料速率来计算的。
详细的聚合方法条件和一些特性列于表5中。调节清除剂供料速率来优化催化剂效率,并且该供料速率在0(没有清除剂)到15μmol/min变化。催化剂供料速率也可以根据系统中杂质水平调节来实现所列的目标转化率。将异己烷用作聚合溶剂,并且它的供料速率是56.7g/min。全部反应在大约2.4mpa/g的压力进行,除非另有提及。用于实施例162-167的聚合方法的另外的加工条件和所生产的聚合物的性能包括在下表5中。
乙烯含量是使用ftir根据astmd3900测定的。
熔体流动速率(mfr)是根据astmd1238使用2.16kg载荷和在230℃温度测定的。高载荷熔体指数(也称作i21)是根据astmd-1238在190℃,在21.6kg载荷下所测量的熔体流动速率。hlmi单位是g/10min或者dg/min。熔体指数比(mir)是高载荷熔体指数与熔体指数的比率,或者i21/i2。
乙烯基链端,乙烯叉基链端和亚乙烯基链端的数目是使用1hnmr,使用1,1,2,2-四氯乙烷-d2作为溶剂在至少400mhznmr分光计上测量的。质子nmr数据是在120℃在5mm探针中,使用varian分光计收集的,其的1h频率是400mhz。数据是使用最大脉冲宽度45°,脉冲间隔5秒和平均120个瞬态的信号来记录的。将光谱信号积分,并且通过不同的基团乘以1000和除以碳总数的结果来计算每1000个碳原子的不饱和度类型数。
链端不饱和度是如下来测量的。感兴趣的乙烯基共振是5.0-5.1ppm(vra),乙烯叉基基共振是4.65-4.85ppm(vdra),亚乙烯基共振是5.31-5.55ppm(vyra),三取代的不饱和物质是5.11-5.30ppm(tsra)和感兴趣的脂肪族区是0-2.1ppm(ia)。
乙烯基数/1000个碳是由式:(vra*500)/((ia+vra+vyra+vdra)/2)+tsra)确定的。同样,乙烯叉基数/1000个碳是由式:(vdra*500)/((ia+vra+vyra+vdra)/2)+tsra)确定的,亚乙烯基数/1000个碳是由式(vyra*500)/((ia+vra+vyra+vdra)/2)25+tsra)确定的,和三取代基团数是由式(tsra*1000)/((ia+vra+vyra+vdra)/2)+tsra)确定的。vra,vdra,vyra,tsra和ia是上面定义的化学偏移区中积分的校正信号强度。乙烯基链端是作为不饱和的聚合物端基的总摩尔数的摩尔百分比来报告的(即,乙烯基链端,乙烯叉基链端,亚乙烯基链端和三取代烯烃链端的总数)。
小振幅振荡剪切(saos):动态剪切熔体流变数据是用advancedrheometricsexpansionsystem(ares),使用平行板(直径=25mm)以动态模式在氮气氛下测量的。对于全部实验,流变仪是在190℃至少30分钟热稳定的,然后将树脂(聚合物组合物)的压塑样品插入到平行板上。为了测定样品的粘弹性行为,在190℃的温度在10%的恒定应变进行0.01-385rad/s的频率扫描。将氮气流循环通过样品烘箱来使得实验过程中扩链或者交联最小化。将正弦剪切应变施加到所述材料。如果应变振幅足够小,则所述材料是线性行为。如本领域技术人员将知晓的,所形成的稳态应力也将以相同频率正弦振荡,但是将相对于应变波偏移相位角δ。所述应力先于应变δ。对于纯弹性材料来说δ=0°(应力是与应变同相的),和对于纯粘性材料,δ=90°(应力先于应变90°,虽然应力是与应变速率同相的)。对于粘弹性材料,0<δ<90。作为频率的函数的复数剪切粘度,损耗模量(g”)和储能模量(g’)是通过小振幅振荡剪切测试来提供的。动态粘度也称作复数粘度或者动态剪切粘度。所述相位角或者损耗角δ是g”(剪切损耗模量)与g'(剪切储能模量)之比的反正切。
差示扫描量热法(对于大规模产物)(dsc-程序-2)。峰值熔点(tm,也称作熔点),峰值结晶温度(tc,也称作结晶温度),玻璃化转变温度(tg),熔化热(hf)和百分比结晶度是使用下面的dsc程序根据astmd3418-03测定的。差示扫描量热法(dsc)数据是使用tainstrumentsq2100型机器来进行的。将称重为大约5-10mg的样品密封在铝密封样品盘中。dsc数据是通过首先以10℃/min的速率将样品逐步加热到200℃来记录的。将该样品保持在200℃2分钟,然后以10℃/min的速率冷却到-70℃,随后等温持续2分钟和以10℃/min加热到200℃。记录该第一和第二循环热事件二者。测量吸热峰下面的面积,并且用于确定熔化热和百分比结晶度。百分比结晶度是使用式:[熔融峰下的面积(j/g)/b(j/g)]*100来计算的,其中b是主要单体组分的100%结晶均聚物的熔化热。用于b的这些值获自polymerhandbook,第四版,由johnwileyandsons出版,纽约1999,但是条件是将189j/g值用作100%结晶聚丙烯的熔化热,将290j/g的值用于100%结晶聚乙烯的熔化热。此处所报告的熔融和结晶温度是在第一冷却/第二加热循环获得的,除非另有指示。
在dsc程序-1和dsc程序-2相矛盾的情况中,应当使用dsc程序-2。
具有三个检测器(gpc-3d)的凝胶渗透色谱
mw,mn和mw/mn是使用高温凝胶渗透色谱(agilentpl-220)测定的,其装备有三个在线检测器,差示射率检测器(dri),光散射(ls)检测器和粘度计。实验细节,包括检测器校正,描述在:t.sun,p.brant,r.r.chance和w.w.graessley,macromolecules,第34卷,第19期,第6812-6820页(2001)以及其中的参考文献中。使用三个agilentplgel10μm混合-bls柱。标称流速是0.5ml/min,和标称注入体积是300μl。各种转移管线,柱,粘度计和差示折射计(dri检测器)容置在保持于145℃的烘箱中。用于实验的溶剂是通过将6g丁基化羟基甲苯作为抗氧化剂溶解在4l的aldrich试剂级1,2,4-三氯苯(tcb)中来制备的。该tcb混合物然后通过0.1μm特氟龙过滤器过滤。该tcb然后用在线脱气机脱气,然后进入gpc-3d。聚合物溶液是通过将干燥聚合物置于玻璃容器中,加入期望量的tcb,然后将该混合物在160℃加热和连续摇动大约2小时来制备的。全部量是重力法测量的。用于表示聚合物浓度的tcb密度(单位质量/体积)在室温是1.463g/ml和在145℃是1.284g/ml。注入浓度是0.5-2.0mg/ml,并且较低的浓度用于较高分子量的样品。在运行每个样品之前,将dri检测器和粘度计吹扫。然后将所述设备中的流速增加到0.5ml/min,和使得dri稳定8小时,然后注入第一样品。ls激光器在运行样品之前至少1-1.5小时打开。在色谱图每个点的浓度c是由减去基线的dri信号idri,使用下面的等式来计算的:
c=kdriidri/(dn/dc)
其中kdri是通过校正dri所确定的常数,和(dn/dc)是系统的折射率增量。对于在145℃和λ=690nm的tcb,折射率n=1.500。本说明书整个gpc-3d中方法的参数单位是这样,即,浓度以g/cm3表示,分子量以g/mol表示,和特性粘度以dl/g表示。
ls检测器是wyatttechnologyhightemperaturedawnheleos。在色谱图每个点处的分子量m是通过使用用于静态光散射的zimm模型分析ls输出来测定的(m.b.huglin,lightscatteringfrompolymersolutions,academicpress,1971):
在此,δr(θ)是在散射角θ所测量的过量的瑞利散射强度,c是由dri分析所测定的聚合物浓度,a2是第二维里系数。p(θ)是用于单分散无规线圈的形状因子,和ko是用于所述系统的光学常数:
其中na是阿佛加德罗常数,和(dn/dc)是所述系统的折射率增量,其采用了与获自dri方法的相同的值。对于在145℃和λ=657nm的tcb,折射率n=1.500。
高温viscotekcorporation粘度计(其具有以惠斯通电桥结构布置的四个毛细管,以及两个压力传感器)用于测定比粘度。一个传感器测量了横跨传感器的总压力降,和另一个(其位于所述桥的两侧之间)测量了压力差。流过粘度计的溶液的比粘度ηs是由它们的输出来计算的。在色谱图每个点的特性粘度[η]是由下面的等式来计算的:
ηs=c[η]+0.3(c[η])2
其中c是浓度,并且是由dri输出测定的。
支化指数(g'vis)是使用gpc-dri-ls-vis方法的输出如下来计算的。样品的平均特性粘度[η]avg是如下来计算的:
其中总和取自积分限度之间的所有色谱切片i。
支化指数g'vis定义为:
mv是粘均分子量,基于通过ls分析所测定的分子量。z平均支化指数(g'zave)是使用ci=在该聚合物峰中在切片i中的聚合物浓度乘以切片质量的平方mi2来计算的。
测试样品的数据加工所用的mark-houwink参数是:1)对于乙烯聚合物:k/a=0.000579/0.695;和2)对于丙烯聚合物:k/a=0.0002288/0.705)。
全部分子量是重量平均的,除非另有指示。全部分子量是以g/mol报告的,除非另有指示。
在gpc-3d程序和“快速gpc”之间矛盾的情况中,应当使用紧上面的gpc-3d程序。关于测定mw,mn,mwd的方法进一步的细节描述在us2006/0173123第24-25页,第[0334]-[0341]段中。
1%正割挠曲模量是使用iso37-类型3样条测量的,并且十字头速度是1.0mm/min和支撑跨度30.0mm,使用instron机器根据astmd790(a,1.0mm/min)测量。
表1:使用未负载的茂金属催化剂的小规模乙烯丙烯共聚。催化剂=0.015μmol,mao=500当量,异己烷溶剂,115psi丙烯,总体积=5ml,tp=70℃。
表2:使用0.39mg负载的催化剂的小规模丙烯聚合和乙烯丙烯共聚。条件:异己烷溶剂,115psi丙烯,tonal=4μmol,总体积=5ml,tp=70℃。
表3:使用未负载的茂金属催化剂的小规模丙烯聚合和乙烯丙烯共聚:催化剂=0.015μmol,mao=500当量,异己烷溶剂,115psi丙烯,总体积=5ml,tp=70℃。
表5:使用mcn14/n,n-二甲基苯胺
如表1和图2所示,在类似条件下的丙烯-乙烯共聚中,具有2-己基取代的mtc1已经表现出高于具有2-环丙基取代的mtc5(对比)和mtc6(对比)以及具有2-ipr取代的mtc7(对比)的mw能力。
如表2和图3所述,具有2-线性烷基(至少4个碳)取代的负载的催化剂a,b,c已经表现出对于丙烯聚合物和从均聚丙烯到低c2(~10wt%c2)到高c2(大约50c2wt%)的共聚物的高mw能力。作为对比,具有2-环丙基取代的对比催化剂d对于均聚丙烯具有中等mw能力,但是对于丙烯-乙烯共聚物非常低的mw能力,具有2-ipr取代的对比催化剂e对于均聚丙烯和低c2(~10wt%c2)共聚物具有低的mw能力,和对于高c2(大约50c2wt%)共聚物具有高mw能力,具有2-me取代的对比催化剂f对于均聚丙烯具有高mw能力,但是对于丙烯-乙烯共聚物仅仅具有中等mw能力。
如表3和图4所示,对于均聚丙烯和丙烯-乙烯共聚物,具有2-丁基取代的mtc4表现出高于具有2-ipr取代的mtc11(对比)的mw能力。
如表4和图5和6所示,对于在类似条件下的丙烯聚合来说,具有4-取代的苯基的mtc1和mtc3表现出高于对比茂金属mtc12和mtc13的ipptm和mw能力。
如表4和图7和8所示,对于在类似条件下的丙烯-乙烯共聚来说,具有4-取代的苯基的mtc1和mtc3表现出高于对比茂金属mtc12和mtc13的mw能力和活性。
令人感兴趣地,如表5和图9所示,本发明的催化剂mtc14不仅表现出在高温(120℃)良好的热稳定性和生产率,而且产生了具有长链支化的聚合物,其是通过g’vis是0.935(实施例163)和0.886(实施例164)来证实的。图10-12所示的流变性测量进一步支持了这个结论。
本文所述的全部文献通过引用并入本文,包括任何优先权文献和/或测试程序,只要它们不与本文相矛盾。如从前述一般描述和具体实施方案中显然可见的,虽然已经显示和描述了本发明的形式,但是在不背离本发明的精神和范围的情况下,可以进行各种改变。因此,不意在由此限定本发明。同样,术语“包含”被认为与术语“包括”是同义的。同样无论何时当组成、要素或者要素的组前面带有连接词“包含”时,应理解为我们还预期了在所述组成、一种要素或多种要素的记载前面带有连接词“基本上由……组成”,“由……组成”,“选自由……组成的组”或“是”的相同的组成或要素组,反之亦然。
- 还没有人留言评论。精彩留言会获得点赞!