用于合成(1R,2S)‑贝达喹啉的手性诱导剂的制作方法
本发明属有机合成技术领域,具体涉及到合成(1R,2S)-贝达喹啉的手性诱导剂。
技术背景
耐多药结核病(multi-drug-resistant tuberculosis,MDR-TB)是一种由细菌引起的,对异烟肼和利福平耐药,以及任何氟喹诺酮类药和任何二线抗结核注射药物(阿米卡星、卡那霉素或卷曲霉素)都有抗药性的结核病。治疗这些形式的结核病,标准的6个月一线抗结核药物不起作用,并可能需要长达两年或更长的治疗时间,而这些药物药效小、毒性大,使用一个疗程标准结核病药物的成本约为20美元,而耐多药结核病药物的成本可以高达5000美元,甚至更高。全球范围内,接受治疗的病人当中,治愈率达50%。调查发现对MDR-TB疗法失败的病人3年内100%死亡,临床迫切需要更好疗效的药物。
2012年12月美国FDA加速批准了强生公司(Johnson & Johnson)用于治疗耐药性结核病的药物富马酸的(1R,2S)-贝达喹啉,结构式为:,
(1R,2S)-富马酸贝达喹啉通过抑制结核分枝杆菌(M.tb)的ATP合成酶来杀死结核分枝杆菌。该药成为近40多年来首个具有全新作用机制的抗TB药物,同时也是有史以来首个明确用于MDR-TB的抗TB药物。其50%治愈率的时间为13周(原联合用药2年),其80%治愈率的时间为6个月。与联合用药(2-4种药物)相比,该药物的治愈率大幅提高,治疗周期大幅缩短。
贝达喹啉有四个光学异构体,只有(1R,2S)构型有效作为药用。原研(1R,2S)-贝达喹啉的合成专利(授权公告号:CN1286371C;CN101180302B)公开了一步合成法,如图1:由6-溴-3-苄基-2-甲氧基喹啉在低温下(-72~-78℃),由二(异丙基)锂铵(LDA)脱苄基的质子后,与3-二甲胺基-1-萘基-1-丙酮加成生成贝达喹啉的4个光学异构体混合物(A为目标产物),反应物料浓缩, 再以乙醇处理得到主导为A与A’的外消旋产物,其中含有少量的B与B’,再经用手性拆分剂(R)-联萘酚磷酸酯的拆分,得到(1R,2S)-贝达喹啉(A),总收率很低,仅7~9%(以6-溴-3-苄基-2-甲氧基喹啉计)。
另外有研究报道,如Shibasaki(Saga Y et al. Catalytic Asymmetric Synthesis of R207910. J. Am Chem. Soc.2010, 132: 7905-7907)和Chandrasekhar(Chandrasekhar S et al. Practical Syntheses of (2S)-R207910 and (2R)-R207910. Eur. J. Org. Chem. 2011, 2057-2061)分别报道了以不对称催化的方法构建第一个碳手性,再以不对称合成构建第二个碳手性。这条路线存在反应步骤多、或收率极低、或使用昂贵的试剂、手性配体、贵金属催化剂等各种问题,其制造成本很高。
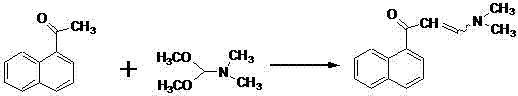
国内也有相关制备专利的申请,专利1(申请号CN105175329A):以-3-溴苄基-6-溴-2-甲氧基喹啉为原料,先与镁制备成格式试剂,再与1-萘甲醛加成等,也是多步骤的反应,关键的是合成产物中4个异构体的比例也未提及;专利2(申请号CN105085396A):为避免3-二甲氨基-1-萘基-1-丙酮在反应中烯醇化而导致加成产物的收率下降,将1-萘乙酮与N,N-二甲基甲酰胺二甲基缩酮反应生成3-二甲氨基-2-(1-萘基)-丙-2-烯-1-酮,如图2。
虽然此萘酮中间体与6-溴-3-苄基-2-甲氧基喹啉在LDA存在下的加成收率有所提高,但增加了高压氢化步骤,且目标光学异构体的比例未有改善;专利3(申请号CN105198808A):则是将拆分后光学异构体废料分解成最初的2个原料,再重复利用。
以上的各种合成方法都各存在不足之处,在原研专利中4个光学异构体混合物的总收率也接近40%,但拆分后(1R,2S)-贝达喹啉的收率仍很低。因此有必要开发更高效的方法来合成(1R,2S)-贝达喹啉。
LDA在手性的邻氨基醇锂存在下,诱导脱质子形成手性碳负离子,或与手性诱导剂形成手性复合物,该负离子参加各种反应,可形成某一个光学异构体占主导的产物(Collum, D.B. et al.J. Am. Chem. Soc.2001,123: 9135–9143; M. Hasegawa et al, Tetrahedron, 2000, 56:10153-10158;H. Pellissier, Tetrahedron, 2007, 63:9267-9331)。在本发明中加入了特定的手性邻氨基醇锂,成功诱导生成了目标对映异构体—(1S,2R)-贝达喹啉作主导的产物。
技术实现要素:
本发明提供了合成(1R,2S)-贝达喹啉的手性诱导剂,该手性诱导可显著提高反应产物中的(1R,2S)-贝达喹啉光学异构体的比例。
为实现上述目的,本发明采用如下技术方案:
首先制备醇锂,通常在低温-72~-78℃下,于手性邻氨基醇的四氢呋喃溶液中加入正丁基锂得到相应的醇锂,再加入二(异丙基)锂铵(LDA),慢慢滴加6-溴-3-苄基-2-甲氧基喹啉的四氢呋喃溶液,得到相应的苄基锂,最后在低温下-72~-78℃加入3-N,N-二甲胺基-1-萘基-1-丙酮的四氢呋喃溶液,生成产物为4个光学异构体的混合物(A与A’;B与B’),反应式如图3。
向反应物料中加饱和氯化铵溶液中止反应,充分沉淀,过滤去除沉淀(主要为B与B’),反应物料浓缩后,以乙醇打浆加热,再冷却得到(1R,2S)-贝达喹啉粗品,该粗品于异丙醇中重结晶得光学纯度与化学纯度高的(1R,2S)-贝达喹啉,其可用于制备富马酸贝达喹啉。
其中,在LDA脱6-溴-3-苄基-2-甲氧基喹啉的苄位质子前,采用了手性邻氨基醇锂作手性诱导剂。该手性邻氨基醇锂由正丁基锂在四氢呋喃溶液中制备,正丁基锂的用量与手性邻氨基醇等当量,稀释手性邻氨基醇的四氢呋喃用量为3ml/g。其它的反应条件、操作及反应后处理方法均参照原研专利(CN101180302B)。
其中,手性邻氨基醇的用量为6-溴-3-苄基-2-甲氧基喹啉的1.0至2.0当量,优选1.10倍当量。
本发明筛选出了一个实用的手性诱导剂及其优选的用量。我们考察了三个常见的、价格较低的手性邻氨基醇:N-甲基-L-脯氨醇(如图4中的1结构式)、N-苄基-L-脯氨醇(如图4中的2结构式)及(1R,2S)-1-苯基-2-(1-吡咯烷基)丙烷-1-醇(如图4中的3结构式)的手性诱导效果:
文献(Collum, D. B. et al.J. Am. Chem. Soc.2001,123: 9135–9143)中手性诱导剂用量,最佳为2.2倍当量,当本发明中底物为6-溴-3-苄基-2-甲氧基喹啉时,其2位甲氧基的氧孤对电子可与锂配位(图5中的1结构式)形成5元环,因此可能减少了手性配基的用量,至1.10当量:
本发明反应中参照原专利方法(CN1286371C),采用高效液相法(HPLC)测定了反应物料中各光学异构体之间的比例。在没有手性诱导剂存在下,两对对映异构体的量A=A’,B=B’;而采用手性邻氨基醇3(图4中的3结构式)诱导后的结果为A/A’ = 92:8,比文献案例(Collum, D.B.; et al.)的效果(50:1)差,或许是因为本案中6-溴-3-苄基-2-甲氧基喹啉的苄位锂盐(图5中的1结构式),其空间位阻远大于2-环丙基-1-丙炔锂 (图5中的2结构式),因此手性诱导效果下降;采用手性邻氨基醇2(图4中的2结构式)诱导结果为A/A’=82:18;采用手性邻氨基醇结构2的诱导结果较差,为A/A’ = 64:36;虽然手性邻氨基醇3的效果最优,但考虑到手性邻氨基醇2的来源更方便,因此手性邻氨基醇2更具有应用价值。
本发明的技术优点:
在没有手性诱导剂存在下,两对非对映体的比例(A+A’)/(B+B’)为4:1。而在本案中,(A+A’)/(B+B’)略有提高,为4.8-5:1。
本发明中无需化学拆分剂,从浓缩后反应物料中直接结晶得到(1R,2S)-贝达喹啉粗品,其中仅含少量的对映体A’(有时也会含有极少量B与B’)。采用N-苄基-L-脯氨醇作手性诱导剂,(1R,2S)贝达喹啉粗品的ee值可达90%。该粗品经异丙醇重结晶得到的(1R,2S)-贝达喹啉,光学纯度ee值99.2%,及化学纯度99.6wt%,(1R,2S)-贝达喹啉达到收率15.6wt%(以6-溴-3-苄基-2-甲氧基喹啉计),其质量标准到达进一步制备药物—(1R,2S)-贝达喹啉富马酸的要求。本发明割除了复杂的化学拆分法及昂贵的拆分剂,收率也大幅提高(从7%~9%提高到15.6%)。
参照原研专利(公开号101547904A),(1R,2S)-贝达喹啉与富马酸成盐结晶,可制备抗结核耐多药的药物—富马酸贝达喹啉。
附图说明
图1为原研专利中(1R,2S)-贝达喹啉的合成反应过程图;
图2为 3-二甲氨基-2-(1-萘基)-丙-2-烯-1-酮的合成反应过程图;
图3为(1R,2S)-贝达喹啉的手性诱导反应图;
图4为三个手性邻氨基醇的结构式;
图5为锂盐结构式。
具体实施方式
为进一步公开而不是限制本发明,以下结合实例对本发明作进一步的详细说明。
(1R,2S)-贝达喹啉的制备
实施例1
在氮气保护下,在干燥的500mL四口玻璃反应瓶中, 加入THF 45ml和N-苄基-L-脯氨醇15.4g(80.5mmol, 1.10eq),将反应瓶置于冷阱中(-72~ -78℃),再加入2.5M正丁基锂的正己烷溶液 32ml(80.5mmol),接着加入2.0M LDA (庚烷-乙苯-四氢呋喃的混合溶剂) 44.0 ml(88.0mmol)、THF 21 ml。将6-溴-3-苄基-2-甲氧基喹啉24.0 g(73.2mmol, 1.0eq)溶于THF 30 ml中,缓慢滴入至上述四口瓶中,在70 min内滴加完毕,加料过程保持内温为-72~-78℃,加毕继续搅拌3 h。在随后的160 min内,缓慢滴加入3-N,N-二甲胺基-1-萘基-1-丙酮17.4 g(76.9mmol)与THF 50 ml配成的溶液,滴加过程保持内温-72~-78℃,加毕,继续反应3h。反应物料的HPLC检测结果:对映体A/A’=82:18(ee:64%);非对映体(A+A’)/(B+B’) = 4.8。在没有手性诱导剂存在下,两对非对映体的比例(A+A’)/(B+B’)为4:1。
在上述反应物中缓慢加入饱和氯化铵水溶液30 mL,保持搅拌缓慢升至室温后,放置12 h,析出沉淀,抽滤,用乙酸乙酯25 mL 浇洗滤饼,滤饼中绝大部分为非对映体(B,B’)。滤液分离出有机层,加入无水硫酸钠干燥后,减压除去溶剂,残余物中加入乙醇120mL,加热至80℃打浆并搅拌2 h。停止加热后自然冷却至室温,继续搅拌2 h。抽滤,用乙醇25 mL 浇洗滤饼。在室温下真空干燥,得贝达喹啉(1R,2S)粗品:类白色固体,10.4g,HPLC检测结果:A/A’=95:5(ee:90%),含2.3wt%的(B+B’)。
上述(1R,2S)-贝达喹啉粗品9.8g,加异丙醇250ml,加热溶解,冷却至0℃结晶,过滤,少量冷异丙醇洗涤,真空干燥得到白色结晶:(1R,2S)-贝达喹啉6.40g,收率15.6wt%(以6-溴-3-苄基-2-甲氧基喹啉计),光学纯度ee:99.2%,化学纯度99.6%(不含B与B’)。
实施例2
在氮气保护下,在干燥的500mL四口玻璃反应瓶中,加入THF 67ml和N-苄基-L-脯氨醇22.4g(117mmol, 1.50eq),将反应瓶置于冷阱中(-72~ -78℃),再加入2.5M正丁基锂的正己烷溶液 46.5ml(117mmol),其它步骤操作及物料用量同实施例1,所得的反应物料HPLC检测结果:对映体A/A’=81:19;非对映体(A+A’)/(B+B’) = 5:1。
实施例3
在氮气保护下,在干燥的500mL四口玻璃反应瓶中,加入THF 90ml和N-苄基-L-脯氨醇30.8g(161mmol,2.0eq),将反应瓶置于冷阱中(-72~ -78℃),再加入2.5M正丁基锂的正己烷溶液 64ml(161mmol),其它步骤及物料用量同实施例1,所得的反应物料HPLC检测结果:对映体A/A’=82:18;非对映体(A+A’)/(B+B’) = 5.1:1。
实施例4
在氮气保护下,在干燥的500mL四口玻璃反应瓶中,加入THF 28ml和N-甲基-L-脯氨醇9.26g(80.5mmol,1.10eq),将反应瓶置于冷阱中(-72~ -78℃),再加入2.5M正丁基锂的正己烷溶液 32ml(80.5mmol),其它步骤及物料用量同实施例1,所得的反应物料HPLC检测结果:对映体A/A’=64:36;非对映体(A+A’)/(B+B’) = 5:1。
实施例5
在氮气保护下,在干燥的500mL四口玻璃反应瓶中,加入THF 49ml和(1R,2S)-1-苯基-2-(1-吡咯烷基)丙烷-1-醇16.5g(80.5mmol,1.10eq),将反应瓶置于冷阱中(-72~ -78℃),再加入2.5M正丁基锂的正己烷溶液 32ml(80.5mmol),其它步骤及物料用量同实施例1,所得的反应物料HPLC检测结果:对映体A/A’=92:8; 非对映体(A+A’)/(B+B’) = 5:1。
后处理过程同实施例1,得到(1R,2S)-贝达喹啉6.81g,收率18.1%(以6-溴-3-苄基-2-甲氧基喹啉计),光学纯度ee:99.5%,化学纯度99.7%(不含B与B’)。
实施例6
在氮气保护下,在干燥的500mL四口玻璃反应瓶中,加入THF 49ml和(1R,2S)-1-苯基-2-(1-吡咯烷基)丙烷-1-醇33.0g(161mmol,2.0eq),将反应瓶置于冷阱中(-72~ -78℃),再加入2.5M正丁基锂的正己烷溶液 32ml(80.5mmol),其它步骤及物料用量同实施例1,所得的反应物料HPLC检测结果:对映体A/A’=92:8;非对映体(A+A’)/(B+B’) = 4.9:1。
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
- 还没有人留言评论。精彩留言会获得点赞!