苯甘氨酸烷基取代类衍生物及其合成方法与流程
本发明属于有机合成化学技术领域,具体涉及一种合成苯甘氨酸烷基取代类衍生物及方法。
背景技术:
苯甘氨酸类衍生物是一类重要的医药中间体,常用于合成氨苄青霉素、头孢氨苄、头孢拉定等β-内酰胺类抗生素,也用于合成多肽激素和多种农药,在生物化学、药物化学等领域具有巨大的发展潜力和广阔的应用前景。
近年来,随着生物体化学的不断深入研究,氨基酸这类小分子生物活性物质逐渐引起人们关注。利用化学合成方法对简单的氨基酸结构进行修饰与改造,得到系列氨基酸类衍生物,从而应用于生物肽或药物研究,具有十分重要的意义。其中,通过过渡金属催化的C-H活化对氨基酸进行官能团化相较于传统的化学合成方法,在一定程度上展现了其高效性、易操作性、原子经济性等优势。
碳氢活化反应从比较容易的芳基化或烷氧化过程发展到可以进行SP3的烷基化过程有了很大的发展,2013年日本Naoto Chatani课题组受Daugulis课题组碳氢活化芳基化的启发,完成了苯环上烷基化的过程。(Yoshinori Aihara and Naoto Chatani, J. Am. Chem. Soc. 2013, 135, 5308−5311)同年,Shi课题组使用8-氨基喹啉为导向基团在直链底物中引入烷基化(Kai Chen,a Fang Hu,b Shuo-Qing Zhanga and Bing-Feng Shi*ab, Chem. Sci., 2013, 4, 3906),同时Chen课题组先后使用8-氨基喹啉和吡啶酰胺为导向基团在直链底物中引入烷基化。(Shu-Yu Zhang, Gang He, William A. Nack, Yingsheng Zhao, Qiong Li, and Gong Chen*, J. Am. Chem. Soc. 2013, 135, 2124−2127.)。2014年,Yu课题组利用C-H 活化反应在直链烷烃上完成烷基化过程。(Ru-Yi Zhu, Jian He, Xiao-Chen Wang, and Jin-Quan Yu*/ J. Am. Chem. Soc. 2014, 136, 13194−13197.)。
技术实现要素:
本发明的目的在于提供一种条件温和、较高产率、绿色环保的合成α-苯甘氨酸烷基取代衍生物及方法。
为了实现上述目的,本发明的技术方案如下:一种苯甘氨酸衍生物,其结构如下所示:
其中,R代表直链烷基,优选乙基、丙基、丁基、辛基。
上述衍生物的合成方法,将α-苯基-α-吡啶酰胺甘氨酸甲酯I、烷基化试剂、醋酸钯、碱加入到叔戊醇溶液中,反应得目标产物。
I
所述反应通式为:
优选地,醋酸钯与α-苯基-α-吡啶酰胺甘氨酸甲酯的的摩尔比为0.05~0.1 equiv.,烷基化试剂与α-苯基-α-吡啶酰胺甘氨酸甲酯的的摩尔比为3.0~5.0 equiv.;碱与α-苯基-α-吡啶酰胺甘氨酸甲酯的的摩尔比为2.0~4.0 equiv.。
优选地,反应时间为24h,反应温度为110℃。
与现有技术相比,本发明的显著效果如下:
(1)采用广泛易得、便宜的D-苯甘氨酸为起始原料,利用过渡金属钯催化的碳氢活化的反应方法,反应条件温和,操作简便,选择性好,避免的常规的C-H的预活化。
(2)首次采用醋酸钯作催化剂参与苯甘氨酸的烷基化衍生物的合成,对苯甘氨酸分子结构进行修饰,合成一系列新的苯甘氨酸烷基取代衍生物结构,在生化、药化研究领域具有应用价值。作为一类重要的医药中间体,目标产物可用于合成氨苄青霉素、头孢氨苄、头孢拉定等β-内酰胺类抗生素,可用于药物筛选工作,也可用于合成多肽激素和多种农药。
(3)本发明的方法适用性广,可放大到克级,只需通过延长反应时间,产率稍有下降。此外,导向官能团吡啶酰胺易除去。
附图说明
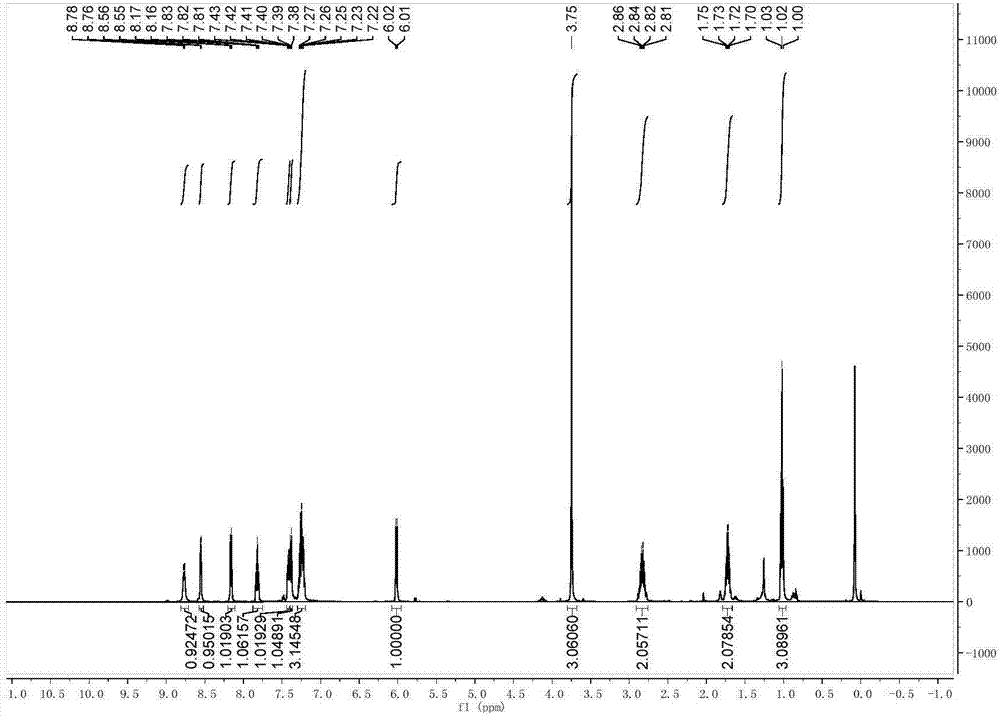
图1为实施例烷基化的核磁共振氢谱(1H NMR)图,其中,图1a为实施例一中产物1a,图1b为实施例二中产物1b,图1c为实施例三中产物1c,图1d为实施例四中产物1d。
图2为实施例烷基化的核磁共振碳谱(13C NMR) 图,其中,图2a为实施例一中产物2a,图2b为实施例二中产物2b,图2c为实施例三中产物2c,图2d为实施例四中产物2d。
实施例一
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘乙烷 (1.0 mmol),醋酸钯 (0.02 mmol),碳酸氢钠 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE+EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状产物α-吡啶酰胺-α-(2-乙基)苯甘氨酸甲酯1a,产率50%。
1H NMR (500 MHz, CDCl3) δ 8.79 (d, J = 7.2 Hz, 1H), 8.55 (d, J = 4.5 Hz, 1H), 8.16 (d, J = 7.7 Hz, 1H), 7.82 (dd, J = 10.3, 5.0 Hz, 1H), 7.46 – 7.35 (m, 2H), 7.26 (ddd, J = 16.9, 11.6, 7.1 Hz, 3H), 6.03 (d, J = 7.6 Hz, 1H), 3.75 (d, J = 1.5 Hz, 3H), 2.98 – 2.86 (m, 2H), 1.32 (t, J = 7.6 Hz, 3H).如图1a。 13C NMR (126 MHz, CDCl3) δ 171.74, 163.86, 149.30, 148.33, 142.90, 137.34, 134.40, 129.43, 128.88, 126.94, 126.70, 126.49, 122.38, 52.79, 52.69, 25.90, 15.51,如图2a。
实施例二
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丙烷 (1.0 mmol),醋酸钯 (0.02 mmol),碳酸氢钠 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状α-吡啶酰胺-α-(2-丙基)苯甘氨酸甲酯产物1b,产率45%。
1H NMR (500 MHz, CDCl3) δ 8.77 (d, J = 7.0 Hz, 1H), 8.55 (d, J = 4.4 Hz, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.82 (t, J = 7.2 Hz, 1H), 7.41 (dd, J = 7.0, 5.2 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.30 – 7.19 (m, 3H), 6.02 (d, J = 7.6 Hz, 1H), 3.75 (s, 3H), 2.83 (dd, J = 18.1, 8.2 Hz, 2H), 1.72 (dd, J = 15.0, 7.5 Hz, 2H), 1.02 (t, J = 7.3 Hz, 3H).如图1b 13C NMR (126 MHz, CDCl3) δ171.75, 163.85, 149.31, 148.32, 141.48, 137.32, 134.53, 130.29, 128.65, 126.95, 126.74, 126.47, 122.38, 52.77, 35.03, 24.51, 14.23,如图2b。
实施例三
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丙烷 (1.0 mmol),醋酸钯 (0.02 mmol),碳酸铯 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状α-吡啶酰胺-α-(2-丙基)苯甘氨酸甲酯产物1b,产率48%。
实施例四
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丙烷 (1.0 mmol),醋酸钯 (0.02 mmol),乙酸铯 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状α-吡啶酰胺-α-(2-丙基)苯甘氨酸甲酯产物1b,产率45%。
实施例五
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丙烷 (1.0 mmol),醋酸钯 (0.02 mmol),乙酸钠 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状α-吡啶酰胺-α-(2-丙基)苯甘氨酸甲酯产物1b,产率47%。
实施例六
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丙烷 (1.0 mmol),醋酸钯 (0.02 mmol),磷酸氢二钾 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状α-吡啶酰胺-α-(2-丙基)苯甘氨酸甲酯产物1b,产率40%。
实施例七
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丙烷 (1.0 mmol),醋酸钯 (0.02 mmol),碳酸钠 (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状α-吡啶酰胺-α-(2-丙基)苯甘氨酸甲酯产物1b,产率44%。
实施例八
向15 mL 耐压管中加入α-苯基-α-吡啶酰胺甘氨酸甲酯 (0.2 mmol),碘丁烷 (1.0 mmol),醋酸钯 (0.02 mmol),base (0.4 mmol), 叔戊醇(1 mL),搅拌下,110℃油浴中加热12~24小时,反应通过薄层色谱TLC 跟踪进程。反应结束后冷却至室温,反应液过短硅胶柱(50% EA/PE-EA),滤液旋蒸除去溶剂,通过柱色谱精细分离,用乙酸乙酯/石油醚洗脱,得到淡黄色油状产物α-吡啶酰胺-α-(2-丁基)苯甘氨酸甲酯1c,产率42%。
- 还没有人留言评论。精彩留言会获得点赞!