二芳基硒醚类衍生物及其制备方法与流程
本发明涉及有机合成领域,且特别涉及一种二芳基硒醚类衍生物及其制备方法。
背景技术:
含有芳基硒醚结构单元的分子广泛应用于医药、农业等领域。一些具有生物活性的含芳基硒醚片段的化合物在医药分子研究中展示了其独特的药理作用。硒类化合物作为一种抗氧化剂是治疗各种癌症、心脏病、炎症的重要化合物,例如硒醚化合物i作为类维生素a(atra)能影响细胞的分化。
随着有机硒化合物在合成中的广泛应用,近年来有机硒化合物在有机合成中的应用得到普遍重视。硒醚类化合物的合成路径引起了无数有机合成工作者的积极思考,并且得出了一些很有效的方法。通过芳环碳与硒原子的c―se偶联是硒醚类化合物合成的主要途径,过渡金属催化被广泛应用于硒醚类化合物的c―se偶联反应。
过渡金属催化合成芳基硒醚的方法中,钯类催化剂较高的毒性、不菲的价格及对含磷配体的依赖制约了其在工业化生产中应用。铜、镍类催化剂也需在加入配体或其它添加剂的情况下才能发挥催化作用,而额外试剂的加入必然也对反应条件及原料分子结构中基团的兼容性起了一定的限制作用。因此,高效性、产率高、具有原子经济性的绿色合成方法在c―se偶联反应制备芳基硒醚类化合物的研究中成为了一个新的导向。
技术实现要素:
本发明的目的在于提供一种二芳基硒醚类衍生物,该二芳基硒醚类衍生物有多环的存在,其结构更加复杂多样,在化工生产、临床医药中也将表现出更加广阔的用途前景。
本发明的另一目的在于提供一种二芳基硒醚类衍生物的制备方法,该方法底物合成简单、试剂比较便宜,并且具有高原子经济性、绿色环保的得到目标分子,给二芳基硒醚类衍生物的工业化生产提供了一条很有价值的途径。
本发明解决其技术问题是采用以下技术方案来实现的:
本发明提出一种二芳基硒醚类衍生物,其结构通式为:
其中,e=co2r,r选自直链烷基、支链烷基、烯基、炔基或者芳香烃基团中任意一种基团;r1选自氢基、卤素基团、直链烷基、支链烷基、酯基或者烷氧基及其衍生物中的任意一种基团;r1可处于苯环的任意位置。
本发明还提供一种二芳基硒醚类衍生物的制备方法,包括以下步骤:
在无水无氧体系中,将二炔类化合物、烃基炔溴、无水乙腈、过渡金属催化剂和有机碱混合并反应得到中间体。在90-100℃条件下,中间体、二苯基二硒醚和甲苯混合反应。
本发明二芳基硒醚类衍生物及其制备方法的有益效果是:本发明提供了一系列新的二芳基硒醚类衍生物相对于普通硒醚衍生物,其有多环的存在,其结构更加复杂多样,在化工生产、临床医药中也将表现出更加广阔的用途前景。本发明的制备方法克服了以往反应中路线过长,底物和反应条件要求苛刻,取代官能团扩展有限的等缺点,该制备方法不但底物合成简单、试剂比较便宜,并且具有高原子经济性、绿色环保的得到目标分子,给二芳基硒醚类衍生物的工业化生产提供了一条很有价值的途径。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,以下将对实施例中所需要使用的附图作简单地介绍。
图1为本发明实施例1制备得到的二芳基硒醚类衍生物的核磁共振氢谱;
图2为本发明实施例1制备得到的二芳基硒醚类衍生物的核磁共振碳谱;
图3为本发明实施例2制备得到的二芳基硒醚类衍生物的核磁共振氢谱;
图4为本发明实施例2制备得到的二芳基硒醚类衍生物的核磁共振碳谱;
图5为本发明实施例3制备得到的二芳基硒醚类衍生物的核磁共振氢谱;
图6为本发明实施例3制备得到的二芳基硒醚类衍生物的核磁共振碳谱。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
在本发明的描述中,需要说明的是,术语“第一”、“第二”等仅用于区分描述,而不能理解为指示或暗示相对重要性。
下面对本发明实施例的二芳基硒醚类衍生物及其制备方法进行具体说明。
本发明实施例提供的一种二芳基硒醚类衍生物,其结构通式为:
其中,e=co2r,r选自直链烷基、支链烷基、烯基、炔基或者芳香烃基团中任意一种基团;r1选自氢基、卤素基团、直链烷基、支链烷基、酯基或者烷氧基及其衍生物中的任意一种基团;r1可处于苯环的任意位置。
本发明实施例中r中的直链烷基选自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基或者其他无支链的烷基中的任意一种基团。优选地r中的直链烷基为乙基。r中的支链烷基选自异丙基、异丁基、叔丁基、异戊基、叔戊基、新戊基、异己基、叔己基、新己基等具有支链的烷基中的任意一种基团。进一步优选地,r中的支链烷基为异丙基。r中的烯基可以是具有支链的烯基也可以是不具有支链的烯基。r中的炔基可以是具有支链的烯基也可以是不具有支链的炔基或者是二者的衍生物。r中的芳香烃基团可以是芳香烷烃也可为芳香烷烃的衍生物或者是二者的衍生物。
在本发明实施例中r1中的卤素基团选自氟基、氯基、溴基或者碘基中任意一种基团。优选地,r1中的卤素基团为氯基团。r1中的直链烷基选自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基或者其他无支链的烷基中的任意一种基团。优选地r1中的直链烷基为乙基。r2中的支链烷基选自异丙基、异丁基、叔丁基、异戊基、叔戊基、新戊基、异己基、叔己基、新己基等具有支链的烷基中的任意一种基团。r2中的烷氧基可以是甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、异戊氧基、叔戊氧基、新戊氧基或者其他的烷氧基团。
本发明实施例还提供一种二芳基硒醚类衍生物的制备方法:
具体的反应式为:
具体的合成包括以下步骤:
s1、制备中间体;
在无水无氧体系中,将二炔类化合物和取代苯乙炔溴加入到无水乙腈中,而后加入过渡金属催化剂和有机碱混合并在室温下搅拌12-20小时反应得到中间体。
采用无水无氧体系是为了保证过渡金属催化剂的活性,若反应体系中存在水或者氧气将会使得过渡金属催化剂失去活性,进而不能合成得到中间体,影响二芳基硒醚类衍生物的生成。而采用过渡金属作为催化剂降低了反应所需的能量,使得反应能够顺利进行。所用的二炔类化合物为碳原子桥连的二炔类化合物。
优选地,过渡金属催化剂为pd(pph3)2cl2和cui,pd(pph3)2cl2和cui的物质的量之比为2.8-3.2:1。采用pd(pph3)2cl2和cui混合作为催化剂时,pd(pph3)2cl2作为催化剂,而cui作为助催化剂,提升催化剂的效能,加快反应速率,提高产率。二者使用的比例保证了催化效果,进一步地加快反应速率,减少副产物。
优选地,有机碱为胺类化合物,例如甲胺、乙胺、丙胺、丁胺、戊胺、二乙胺、二丙胺、二丁胺、二戊胺、二己胺、三乙胺等。进一步优选地有机碱为三乙胺。三乙胺可以反应生成的溴化氢反应,进而促进整个反应平衡向生成物方向进行。
进一步优选地,二炔类化合物、烃基炔溴、过渡金属催化剂和有机碱的物质的量之比为1:2-3:0.03-0.05:2-5。无水乙腈作为溶剂时,二炔类化合物在无水乙腈中的浓度为0.5-0.8mol/l。在该优选值范围内进行的反应速率快,产率高,副反应少。
s2、纯化中间体;
纯化中间体,是去除中间体内可能含有的杂质,减少杂质对后续反应的影响,同时减少二芳基硒醚类衍生物内可能含有的杂质。甚至中间体内含有的杂质可能导致后续不能反应生成得到二芳基硒醚类衍生物。
纯化是将二炔类化合物、烃基炔溴、无水乙腈、过渡金属催化剂和有机碱混合并反应得到的第一反应液首先采用质量百分比为15%的盐酸溶液洗涤去除有机碱,而后再用质量百分比为10%的碳酸氢钠溶液洗涤去除盐酸溶液中的盐酸,再用饱和食盐水洗涤以去除碳酸氢钠。接着采用乙酸乙酯萃取洗涤后第一反应液,除去水相。然后使用无水硫酸镁干燥萃取后得到的有机相。干燥后使用旋转蒸发仪旋蒸以去除有机相中的有机溶剂。最后再用柱层析纯化旋蒸后得到物质,得到纯度高的中间体。其中,柱层析采用的淋洗剂为乙酸乙酯与石油醚的混合液,混合液中乙酸乙酯与石油醚的体积比为1:40-60。采用上述的淋洗液能够良好的分离中间体和杂质。
s3、合成得到二芳基硒醚类衍生物粗品;
在90-100℃条件下,将所述中间体和二苯基二硒醚在甲苯溶剂中搅拌混合反应18-24小时,得到二芳基硒醚类衍生物粗品。温度为影响该反应的重要因素,若温度高于或低于该温度范围则生成物质的结构将发生变化,得不到本发明所需的二芳基硒醚类衍生物。
优选地,中间体与二苯基二硒醚的物质的量之比为1:0.5-1。甲苯作为溶剂时,中间体在甲苯中的浓度是0.1-0.3mol/l。在该优选值范围内进行的反应速率快,产率高、副产物少。
s4、纯化二芳基硒醚类衍生物粗品;
反应得到的二芳基硒醚类衍生物粗品内含有杂质,这些杂质会影响对二芳基硒醚类衍生物结构的鉴定或者影响后续的使用。因此,需要对其进行进一步地纯化。
将制备得到的二芳基硒醚类衍生物粗品即第二反应液经过去离子水洗涤后,用乙酸乙酯萃取,用无水硫酸镁干燥萃取液。过滤后,用旋转蒸发仪蒸干萃取液中溶剂。二芳基硒醚类衍生物粗品用柱层析的方法分体提纯,柱层析的淋洗剂为乙酸乙酯与石油醚的混合液,混合液中乙酸乙酯与石油醚的体积比为1:40-80,柱层析产率约为60%。
本发明实施例制备得到的二芳基硒醚类衍生物的结构通过1hnmr和13cnmr来测定。
本发明实施例提供的一系列新的二芳基硒醚类衍生物相对于普通硒醚衍生物有多环的存在,其结构更加复杂多样。并且,本发明实施例提供的制备方法简便、高效、绿色环保,在化工生产,给二芳基硒醚类衍生物的工业化生产提供了一条很有价值的途径。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1
本实施例提供的一种二芳基硒醚类衍生物,其结构式如(i)式所示。
本实施例还提供一种制备上述二芳基硒醚类衍生物的方法:
将丙二酸二乙酯桥连的二炔化合物(20mmol)与苯乙炔溴(48mmol)加入到反应瓶中,混合在pd(pph3)2cl2和cui作为催化剂的无水无氧催化体系中,其中催化剂的量为0.8mmol,pd(pph3)2cl2:cui=3:1。以三乙胺(50mmol)作碱,以无水乙腈(40ml)为溶剂,室温下搅拌反应12小时,将反应产物先后用15%的盐酸溶液、10%的碳酸氢钠溶液、饱和食盐水分别洗涤;然后用乙酸乙酯萃取,减压旋干,柱层析(淋洗剂中乙酸乙酯与石油醚的体积比为1:40)得到浅棕色固体产物。
在95℃的条件下,将中间体(1mmol)与二苯基二硒醚(0.8mmol)加入到5毫升甲苯溶剂中,反应20小时,得到二芳基硒醚类衍生物粗品。
将二芳基硒醚类衍生物粗品加水洗涤后,用乙酸乙酯萃取,减压旋干,柱层析(淋洗剂中乙酸乙酯与石油醚的体积比为1:50)分离可得到纯化的目标产物,即二芳基硒醚类衍生物,柱层析产率约为62%。
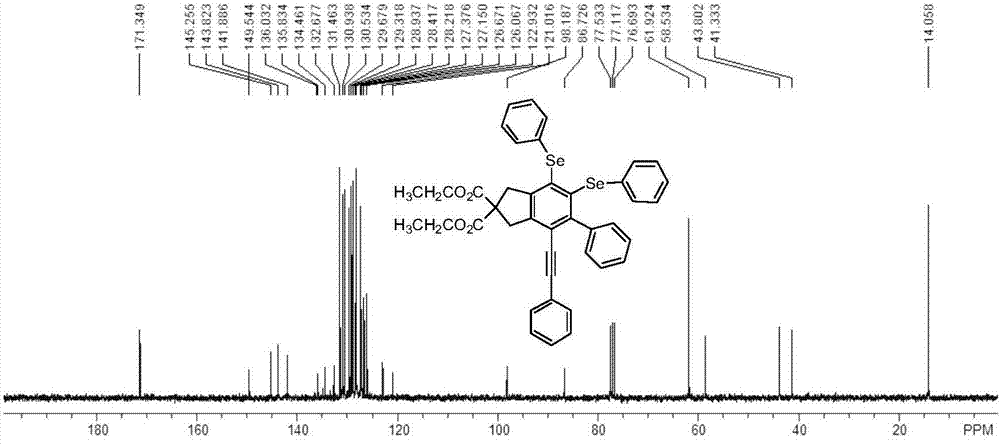
二芳基硒醚类衍生物的结构通过1hnmr和13cnmr来测定,具体图谱见图1和图2。
棕色固体产物:
1hnmr(300mhz,cdcl3):δ7.30―7.27(m,4h),7.23―7.14(m,9h),7.09―7.06(m,5h),7.00―6.97(m,2h),4.20―4.13(q,4h,j=6.9hz),3.80(s,2h),3.44(s,2h),1.23―1.18(t,6h,j=6.9hz);
13cnmr(75.5mhz,cdcl3):δ171.3,149.5,145.3,143.8,141.9,136.0,135.8,134.5,132.7,131.5,130.9,130.5,129.7,129.3,128.9,128.4,128.2,127.4,127.2,126.7,126.1,122.9,121.0,98.2,86.7,61.9,58.5,43.8,41.3,14.1。
实施例2:
本实施例提供的一种二芳基硒醚类衍生物,其结构式如(ii)式所示。
本实施例还提供一种制备上述二芳基硒醚类衍生物的方法:
将丙二酸二乙酯桥连的二炔化合物(20mmol)与对乙基苯乙炔溴(40mmol)加入到反应瓶中,混合在pd(pph3)2cl2和cui作为催化剂的无水无氧催化体系中,其中催化剂的量为0.6mmol,pd(pph3)2cl2:cui=3.2:1。以三乙胺(100mmol)作碱,以无水乙腈(25ml)为溶剂,室温下搅拌反应20小时,将反应产物先后用15%的盐酸溶液、10%的碳酸氢钠溶液、饱和食盐水分别洗涤;然后用乙酸乙酯萃取,减压旋干,柱层析(淋洗剂中乙酸乙酯与石油醚的体积比为1:50)得到浅棕色固体产物。
在90℃的条件下,将中间体(1mmol)与二苯基二硒醚(1.0mmol)加入到10毫升甲苯溶剂中,反应24小时,得到二芳基硒醚类衍生物粗品。
将二芳基硒醚类衍生物粗品加水洗涤后,用乙酸乙酯萃取,减压旋干,柱层析(淋洗剂中乙酸乙酯与石油醚的体积比为1:40)分离可得到纯化的目标产物,即二芳基硒醚类衍生物,柱层析产率约为64%。
二芳基硒醚类衍生物的结构通过1hnmr和13cnmr来测定,具体检测图谱见图3和图4。
棕色固体产物:
1hnmr(300mhz,cdcl3):δ7.23―7.17(m,5h),7.10―7.05(m,8h),7.03―6.97(m,5h),4.17―4.14(q,4h,j=6.0hz),3.79(s,2h),3.42(s,2h),2.71―2.55(m,4h),1.30―1.16(m,12h);
13cnmr(75.5mhz,cdcl3):δ171.4,149.6,145.1,144.9,143.5,143.0,139.4,136.1,135.3,134.6,132.8,131.5,130.8,130.5,129.6,129.3,128.9,127.8,126.9,126.6,126.0,121.4,120.2,98.5,86.4,61.9,58.5,43.8,41.3,28.9,28.9,16.0,15.5,14.1。
实施例3
本实施例提供的一种二芳基硒醚类衍生物,其结构式如(iii)式所示。
本实施例还提供一种制备上述二芳基硒醚类衍生物的方法:
将丙二酸二异丙酯桥连的二炔化合物(20mmol)与对氯苯乙炔溴(60mmol)加入到反应瓶中,混合在pd(pph3)2cl2和cui作为催化剂的无水无氧催化体系中,其中催化剂的量为1.0mmol,pd(pph3)2cl2:cui=2.8:1。以三乙胺(40mmol)作碱,以无水乙腈(33ml)为溶剂,室温下搅拌反应16小时,将反应产物先后用15%的盐酸溶液、10%的碳酸氢钠溶液、饱和食盐水分别洗涤;然后用乙酸乙酯萃取,减压旋干,柱层析(淋洗剂中乙酸乙酯与石油醚的体积比为1:60)得到浅棕色固体产物。
在100℃的条件下,中间体(1mmol)与二苯基二硒醚(0.5mmol)加入到3.5毫升甲苯溶剂中,反应18小时,得到二芳基硒醚类衍生物粗品。
将二芳基硒醚类衍生物粗品加水洗涤后,用乙酸乙酯萃取,减压旋干,柱层析(淋洗剂中乙酸乙酯与石油醚的体积比为1:80)分离可得到纯化的目标产物,即二芳基硒醚类衍生物,柱层析产率约为56%。
二芳基硒醚类衍生物的结构通过1hnmr和13cnmr来测定,具体的检测图谱见图5和图6。
棕色固体产物:
1hnmr(300mhz,cdcl3):δ7.30―7.27(m,2h),7.23―7.18(m,7h),7.07―6.88(m,7h),6.95―6.92(m,2h),5.05―4.96(m,2h),3.73(s,2h),3.42(s,2h),1.23―1.15(m,12h);
13cnmr(75.5mhz,cdcl3):δ170.8,147.9,145.6,143.9,140.0,136.3,136.0,134.6,134.0,133.0,132.6,132.4,131.2,130.8,129.4,128.9,128.7,127.5,126.9,126.3,121.2,120.7,112.4,97.1,87.4,69.5,58.5,43.9,41.2,21.5。
综上所述,本发明实施例1-3提供的一系列新的二芳基硒醚类衍生物相对于普通硒醚衍生物,本发明制备的二芳基硒醚类衍生物有多环的存在,其结构更加复杂多样,在化工生产、临床医药中也将表现出更加广阔的用途前景。本发明的制备方法克服了以往反应中路线过长,底物和反应条件要求苛刻,取代官能团扩展有限的等缺点,该制备方法不但底物合成简单、试剂比较便宜,并且具有高原子经济性、绿色环保的得到目标分子,给二芳基硒醚类衍生物的工业化生产提供了一条很有价值的途径。
以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!