一种同时沉默烟草植物中2个目的基因的方法与流程
本发明涉及植物体内基因沉默技术,特别是涉及通过病毒诱导的基因沉默技术来同时沉默植物体内的2个目的基因技术。
背景技术:
人们经常需要分析植物中基因的功能,有时候是某单个基因的功能,有时候是要同时分析2个或2个以上的基因的功能,这就需要同时敲除2个或2个以上的基因,通常需要先通过遗传转化获得单个基因缺失的突变体,然后在通过遗传杂交筛选双基因缺失的突变体。但在筛选双基因缺失的突变体的过程中,耗费的时间周期长,成本高,工作量非常大,操作复杂,成功率较低。并且有时候同时敲除双基因时会造成植物致死,使得我们不能进一步来研究基因的功能。
病毒诱导的基因沉默(virusinducedgenesilencing,vigs)是近年来在植物中发现的一种转录后基因沉默(post-transcriptionalgenesilencing,ptgs)现象,可引起植物内源mrna序列特异性降解。当携带植物功能基因cdna的病毒侵染植物体后可以诱导植物体触动rna降解机制,使植物体同源基因的mrna被降解,从而可以通过植物表型或生理生化指标上的变化来反映该基因的功能。
利用vigs研究植物基因组功能具有以下优势:1)周期性短、成本较低、构建简易,一般基因在3周以内都能达到沉默的效果;2)不需要遗传转化获得转基因植物即可抑制基因表达,在当代就可以观察基因沉默表型;3)克服基因家族功能冗余的缺点;4)快速比较不同物种之间的基因功能。因此,vigs是植物中分析基因功能非常快速实用的技术。已有大量的报道表明vigs可以应用在植物体内沉默单个基因,如已成功在本生烟中沉默单个基因nbpds、nbics1、nbnpr1、nbsabp2等。然而利用vigs同时沉默2个基因还很少报道。鉴于利用vigs研究植物基因组功能具有多重优势,因此本研究通过借助vigs技术来发明一种同时沉默2个植物中目的基因的方法。
技术实现要素:
为解决上述问题,本发明提供了一种同时沉默烟草植物中2个目的基因的方法。
为实现上述目的,本发明采取的技术方案为:
一种同时沉默烟草植物中2个目的基因的方法,包括如下步骤:
(1)分别采用nbsabp2和nbnac1基因的特异引物,通过rt-pcr在本生烟中分别扩增出nbsabp2基因片段和nbnac1基因片段;分别回收扩增的nbsabp2和nbnac1基因片段;所述nbsabp2基因的特异引物为nbsabp2-f和nbsabp2-r,其序列分别如seqidno.1和seqidno.2所示;nbnac1基因的特异引物是nbnac1-f和nbnac1-r,其序列分别如seqidno.3和seqidno.4所示;
(2)以回收的nbsabp2和nbnac1基因片段为共同模板,利用nbsabp2-f和nbnac1-r为引物,扩增(nbsabp2+nbnac1);回收(nbsabp2+nbnac1)片段;
(3)将回收的(nbsabp2+nbnac1)片段与
(4)将上述的连接产物通过热激的方法转化到大肠杆菌中,获得p(nbsabp2+nbnac1)重组质粒,
(5)利用菌液pcr和质粒pcr来对p(nbsabp2+nbnac1)重组质粒进行鉴定。
(6)将(nbsabp2+nbnac1)克隆到ptrv2(可从biovector质粒载体菌种细胞基因保藏中心购买)载体上,构建病毒重组质粒ptrv2-(nbsabp2+nbnac1);
(7)将构建的病毒重组质粒ptrv2-(nbsabp2+nbnac1)转化到农杆菌gv2260中。
(8)通过利用菌液pcr和质粒pcr来对ptrv2-(nbsabp2+nbnac1)重组质粒进行鉴定。
(9)将ptrv1(可从biovector质粒载体菌种细胞基因保藏中心购买)的农杆菌和带有目的片段ptrv2-(nbsabp2+nbnac1)农杆菌按照1:1的比例混合;用无针头的1ml无菌注射器将混合农杆菌悬浮液注入本生烟烟草下位叶片中;
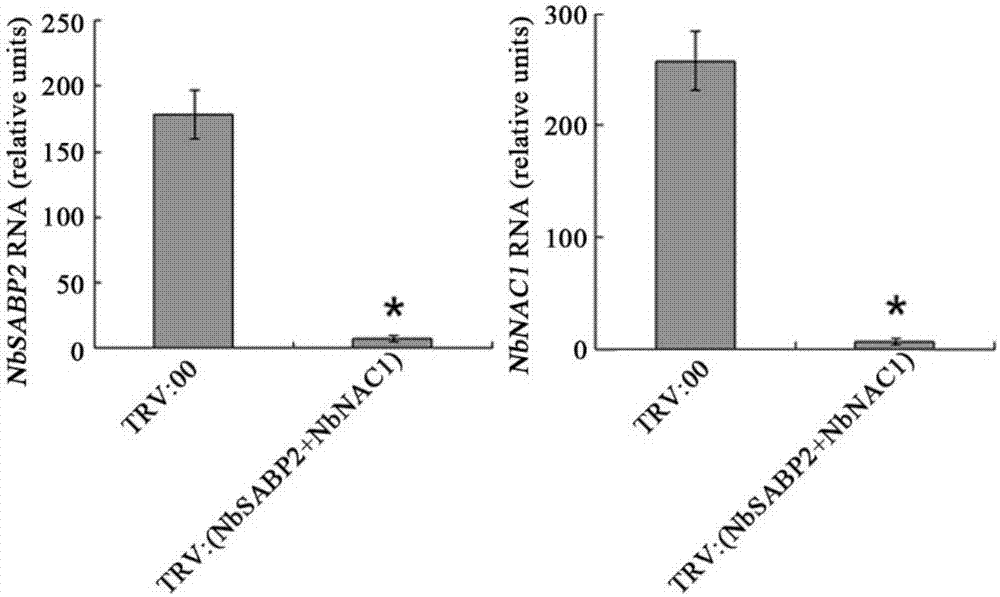
(10)从侵染了12天的农杆菌的本生烟中提取总rna。通过荧光定量pcr的方法检测nbsabp2和nbnac1基因的表达。结果表明nbsabp2和nbnac1基因的表达显著降低。
所述nbsabp2和nbnac1基因的特异引物可由本领域技术人员进行设计,本发明中推荐使用如下:所述nbsabp2基因的特异引物nbsabp2-f和nbsabp2-r,其序列分别如seqidno.1和seqidno.2所示;所述nbnac1基因的特异引物nbnac1-f和nbnac1-r,其序列分别如seqidno.3和seqidno.4所示。
采用本发明的方法,结果表明nbsabp2和nbnac1基因的表达显著降低。实现了同时沉默烟草植物中2个目的基因。
附图说明
图1为本发明实施例一种同时沉默烟草植物中2个目的基因的方法的流程框图。
图2为本发明实施例中通过荧光定量pcr的方法检测nbsabp2和nbnac1基因的表达。
具体实施方式
为了使本发明的目的及优点更加清晰,以下结合实施例对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例一
如图1所示,本发明实施例提供了一种同时沉默烟草植物中2个目的基因的方法,包括如下步骤:
1.分别设计nbsabp2和nbnac1基因的特异引物,注意设计引物时应扩增靠近基因3′区域的片段,引物如下:
nbsabp2-f(seqidno.1):5′-actcttcctttgttttag-3′
nbsabp2-r(seqidno.2):
5′-atcgattgtggccgcggtggatgggcaatctccaagaga-3′
nbnac1-f(seqidno.3):
5′-tctcttggagattgcccatccaccgcggccacaatcgat-3′
nbnac1-r(seqidno.4):5′-ttagtaaggtttctgcatg-3′
值得注意的是为了将nbsabp2和nbnac1基因连接在一起,我们将采用重叠pcr来将它们连接在一起。因此我们在设计引物时要做一些改变,仔细观察上面的引物可以发现nbsabp2-r和nbnac1-f这两条引物要比另外两条引物长很多,那是因为我们在设计引物的时候在nbsabp2-r的3′端加入了20个nbnac1基因5′端的序列,在nbnac1-f的5′端加入19个nbsabp2基因3′端的序列。
2.选取本生烟较嫩叶片为材料,提取本生烟植物的总rna。cdna的合成参照invitrogen公司的反转录体系进行,以oligo(dt)18为通用引物,在37℃合成cdna第一条链。以nbsabp2-f和nbsabp2-r为引物从本生烟中克隆出nbsabp2基因的部分片段,以nbnac1-f和nbnac1-r为引物从本生烟中克隆出nbnac1基因的部分片段,通过rt-pcr在本生烟中分别扩增出大约420bp的nbsabp2基因片段和大约300bp的nbnac1基因片段。
3.利用琼脂糖凝胶回收试剂盒(天根)分别回收扩增的nbsabp2和nbnac1基因片段。
4.以回收的nbsabp2和nbnac1基因片段为共同模板,nbsabp2-f和nbnac1-r为引物,扩增(nbsabp2+nbnac1),这样我们就利用重叠pcr的方法将(nbsabp2+nbnac1)拼接起来了。
5.利用琼脂糖凝胶回收试剂盒(天根)回收(nbsabp2+nbnac1)片段。
6.按照
然后轻轻混匀,在室温条件下反应1h。
7.转化大肠杆菌
将上述的连接产物(2-4μl)在无菌条件下加入到装有100μl大肠杆菌感受态细胞的ep管中,并轻轻摇匀,冰上放置30min;
在42℃条件下水浴中热激60-90s,热激完将ep管放置于冰上冷却3~5min;
然后向ep管中加入300-500μl的液体lb培养基,并将其放入摇床中在37℃条件振荡培养lh左右。
将上述的菌液涂布于含有50mg/l壮观霉素的固体lb培养基上进行筛选,37℃条件下进行过夜培养,大约12-16h。
8.重组质粒的鉴定
挑取以上培养的单菌落,在无菌条件下接种于含有50mg/l壮观霉素的lb液体培养基中,37℃条件下摇床振荡培养12-16h。培养结束后可以通过菌液pcr检测,来鉴定该(nbsabp2+nbnac1)片段是否成功连接到载体上。为了进一步验证,可以从菌液中提取质粒来进行质粒pcr检测,质粒的提取方法可以参照质粒提取试剂盒的操作说明书的步骤进行。
9.病毒重组质粒的构建
按照
在1.5mlep管中依次加入以下成分,具体如下表。
混匀后室温静置5min。
加入2-3μllrclonasetmiienzymemix到样品中,并轻轻震荡混匀。
室温(25℃)条件下反应1-2h。
加入1μlproteinasek溶液到样品中,并轻轻震荡混匀,37℃水浴条件下放置10min,终止反应。
10.重组质粒转化农杆菌gv2260
将1μl的重组质粒加入到装有50μl的农杆菌gv2260感受态细胞的ep管中,轻轻混匀,冰上放置30min。
将ep管放入液氮中冷冻1min后,立即放在37℃水浴锅中水浴3-5min。
然后向ep管中加入400μllb液体培养基,放入摇床中,控温28℃,转速220rpm的条件下振荡培养4~5h。
将菌液涂布在含有四种抗生素(amp、strep、kana、rif)的lb固体培养基上,28℃倒置培养2天,进行筛选。获得ptrv2-(nbsabp2+nbnac1)的重组菌落。
11.重组质粒的pcr鉴定
挑取单菌落放置4mllb液体培养基中,控温28℃,转速220rpm的条件下振荡培养。菌液用于菌液pcr检测。此外,还收集菌体,按质粒提取试剂盒方法提取质粒,进行质粒pcr鉴定。
12.农杆菌侵染
分别挑取ptrv1(可从biovector质粒载体菌种细胞基因保藏中心购买)、ptrv2-(nbsabp2+nbnac1)的重组菌落接入5mllb液体培养基,并加入相应的抗生素,控温28℃,转速220rpm的条件下振荡培养。
然后将活化的菌液接入到上述抗性相同的50mllb液体培养基,另外再加入10mm2-n-吗啉基乙磺酸(mes)和20μm乙酰丁香酮(as),控温28℃,转速220rpm的条件下振荡培养。
在3000rpm的条件下离心10min收集农杆菌菌体,用侵染缓冲液(10mmmgcl2,10mmmes,和200μmas)悬浮菌体,在600nm波长下调节od600值至0.8左右,室温静置3h左右。
将ptrv1的农杆菌和ptrv2-(nbsabp2+nbnac1)农杆菌按照1:1的比例混合。用无针头的1ml无菌注射器将农杆菌悬浮液注入本生烟烟草下位叶片中。
13.荧光定量pcr检测nbsabp2和nbnac1基因的表达
为了避免载体上连接的基因片段的干扰,我们在设计引物时选择靠近基因5’端的序列来设计。引物如下:
nbsabp2-f′(seqidno.5):5′-caggccataaggttac-3′
nbsabp2-r′(seqidno.6):5′-tatcaggcatgaaagc-3′
nbnac1-f′(seqidno.7):5′-acagaaatcagcagcaa-3′
nbnac1-r′(seqidno.8):5′-aatcatcaagcctcaagt-3′
从侵染了12天的农杆菌的本生烟中提取总rna。通过荧光定量pcr的方法检测nbsabp2和nbnac1基因的表达。如图2结果表明nbsabp2和nbnac1基因的表达显著降低。
sequencelisting
<110>扬州大学
<120>一种同时沉默烟草植物中2个目的基因的方法
<130>
<160>8
<170>patentinversion3.3
<210>1
<211>18
<212>dna
<213>人工序列
<400>1
actcttcctttgttttag18
<210>2
<211>39
<212>dna
<213>人工序列
<400>2
atcgattgtggccgcggtggatgggcaatctccaagaga39
<210>3
<211>39
<212>dna
<213>人工序列
<400>3
tctcttggagattgcccatccaccgcggccacaatcgat39
<210>4
<211>19
<212>dna
<213>人工序列
<400>4
ttagtaaggtttctgcatg19
<210>5
<211>16
<212>dna
<213>人工序列
<400>5
caggccataaggttac16
<210>6
<211>16
<212>dna
<213>人工序列
<400>6
tatcaggcatgaaagc16
<210>7
<211>17
<212>dna
<213>人工序列
<400>7
acagaaatcagcagcaa17
<210>8
<211>18
<212>dna
<213>人工序列
<400>8
aatcatcaagcctcaagt18
- 还没有人留言评论。精彩留言会获得点赞!