一种从顶花板凳果中提取东莨菪亭的方法与流程
本发明属于天然产物提取技术领域,具体涉及一种从顶花板凳果中提取东莨菪亭的方法。
背景技术:
顶花板凳果(pachysandraterminalissieb.etzucc.)为黄杨科板凳果属植物,又名顶蕊三角咪、长青草、捆仙七、转筋草和富贵草。该植物具有清热解毒、祛风除湿、舒筋活络、镇静安神的功效,故陕西民间主要用于治疗风湿麻痹、肢体麻木、跌打损伤、月经过多、烦躁不安等,也可作为复方药治疗老年慢性支气管炎、风湿性关节炎等疾病。迄今为止,国内外对顶花板凳果的研究大多仅限于其甾体生物碱成分及三萜类成分的报道,未见对该植物香豆素类化合物提取分离方法的报道。
东莨菪亭在植物中分布广泛,其中在茄科植物东莨菪(scopoliajaponicamaxim)中含量较高。东莨菪亭为香豆素类化合物,其化学名为:7-羟基-6-甲氧基香豆素。东莨菪亭的结构式为:
现代药理学研究表明,东莨菪亭有明显的药理作用,东莨菪亭不仅能够抑制主动脉收缩,缓解动脉粥样硬化所导致的心肌缺血,还能够对抗由高尿酸血症引起的痛风性关节炎。具有抗癌、抗氧化、抗关节炎和降温等临床药理作用;另外,也有对东莨菪亭代谢方面的报道。但是目前提取东莨菪亭的方法所用的溶剂不够环保,对人体有毒害,且成本较高,不够经济。
技术实现要素:
本发明的目的在于提供一种从顶花板凳果中提取东莨菪亭的方法,该方法成本低廉,溶剂环保,操作简单,提取得到的东莨菪亭纯度高。
本发明是通过以下技术方案来实现:
包括以下步骤:
1)将顶花板凳果粉末进行提取处理,得到顶花板凳果乙酸乙酯部位浸膏;
2)取顶花板凳果乙酸乙酯部位浸膏,上样于硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,进行梯度洗脱,对流出液进行检测,将石油醚:乙酸乙酯体积比为(25:1)~(10:1)洗脱的馏分合并,蒸干溶剂,得到第一次过柱部分;
3)取第一次过柱部分,上样于硅胶柱,以石油醚-乙酸乙酯-甲酸系统为洗脱液,进行梯度洗脱,对流出液进行检测,合并主斑点相同的馏分后,蒸干溶剂,得到第二次过柱部分;
4)将第二次过柱部分,上样于硅胶柱,以石油醚-乙酸乙酯-甲酸系统为洗脱液,进行梯度洗脱,对流出液进行检测,蒸干溶剂,得到东莨菪亭。
进一步地,步骤1)中顶花板凳果乙酸乙酯部位浸膏的制备,具体包括以下步骤:
(1)将顶花板凳果粉末用8~15倍量体积分数为95%的乙醇溶液回流提取,回收提取液后浓缩得浸膏;
(2)将浸膏用8~15倍量体积分数为1~2%的盐酸溶解后过滤,得到滤渣;
(3)将滤渣用8~15倍量乙酸乙酯溶解后过滤,回收滤液并浓缩得乙酸乙酯部位浸膏。
进一步地,步骤(1)所述的用体积分数为95%的乙醇回流提取次数为2~4次,每次2~3h;步骤(3)用乙酸乙酯溶解过滤的重复次数为2~4次,每次1~2h。
进一步地,步骤2)的洗脱液中石油醚和乙酸乙酯的体积比为(200:1)~(0:1)。
进一步地,步骤3)的洗脱液中石油醚和乙酸乙酯的体积比为(25:1)~(1:3)。
进一步地,步骤3)将石油醚和乙酸乙酯的体积比为(2:1)~(1:1)洗脱的馏分合并。
进一步地,步骤4)的洗脱液中石油醚和乙酸乙酯的体积比为(5:1)~(1:1)。
进一步地,步骤4)将石油醚和乙酸乙酯的体积比为3:1洗脱的馏分合并。
进一步地,步骤3)和步骤4)的洗脱液中,石油醚和乙酸乙酯的总体积与甲酸的体积之比均为100:(0.1~0.4)。
进一步地,硅胶柱中所用硅胶的粒度均为200~300目;对流出液进行检测是采用薄层色谱法进行检测,于365nm紫外灯下进行检视。
与现有技术相比,本发明具有以下有益的技术效果:
本发明方法将顶花板凳果乙酸乙酯部位浸膏首先过硅胶柱进行第一次梯度洗脱,收集的相同馏分再经反复硅胶柱的梯度洗脱,分离得到东莨菪亭,是首次从中药材顶花板凳果中分离得到单体化合物东莨菪亭的方法。本发明方法简单易行、成本低廉,分离得到的东莨菪亭的纯度达到99%以上。本发明在第一次硅胶柱分离之后使用含甲酸的石油醚-乙酸乙酯系统为流动相,该流动相体系成本低廉、环保低毒,不仅大大降低了环境污染及对人体的毒性伤害,而且节约了成本;更重要的是,在流动相中加入甲酸,不仅能够提高分离效率,而且有效避免了东莨菪亭的开环分解和异构化,更加改善了分离效果。
进一步地,本发明提取过程简单,溶剂环保低毒,且保证提取效率。
进一步地,本发明中甲酸的用量在1%~4%,有效防止影响洗脱液极性,避免导致目标化合物过早地被洗脱出,出现不能成功与其他极性相近的化合物分离的现象。
附图说明
图1为本发明分离得到的东莨菪亭的1h-nmr图谱(400mhz);
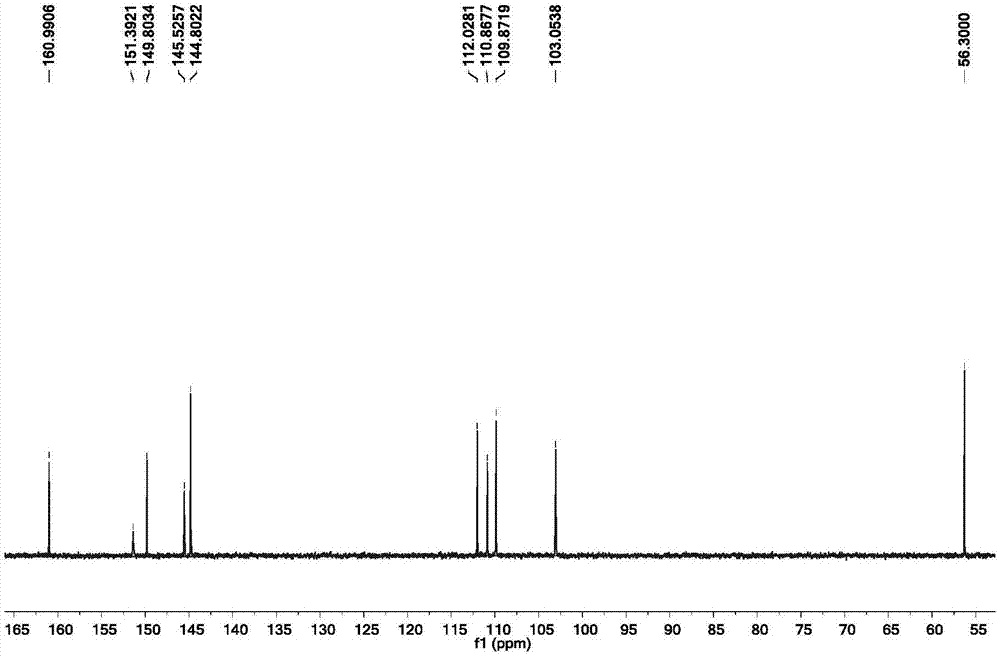
图2为本发明分离得到的东莨菪亭的13c-nmr图谱(100mhz);
具体实施方式
下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
本发明包括以下步骤:
1)制备顶花板凳果乙酸乙酯部位浸膏;具体包括以下步骤:
(1)将顶花板凳果粉末用8~15倍量体积分数为95%的乙醇溶液回流提取,提取次数为2~4次,每次2~3h,回收提取液后浓缩得浸膏;其中倍量均是所加溶液体积相对原料质量的倍数,此处是指95%的乙醇溶液所加的体积是顶花板凳果粉末质量的8~15倍;
(2)将浸膏用8~15倍量体积分数为1~2%的盐酸溶解后过滤,得到滤渣;
(3)将滤渣用8~15倍量乙酸乙酯溶解后过滤,重复次数为2~4次,每次1~2h,回收滤液并浓缩得乙酸乙酯部位浸膏。
2)取顶花板凳果乙酸乙酯部位浸膏,上样于硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,按石油醚:乙酸乙酯=(200:1)~(0:1)的体积比进行梯度洗脱,对流出液进行检测,将石油醚:乙酸乙酯=(25:1)~(10:1)的体积比洗脱的馏分合并,蒸干溶剂,得到第一次过柱部分;
3)取第一次过柱部分,上样于硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,按石油醚:乙酸乙酯=(25:1)~(1:3)的体积比进行梯度洗脱,对流出液进行检测,合并主斑点相同的馏分,即将石油醚:乙酸乙酯=(2:1)~(1:1)的体积比洗脱的馏分合并,蒸干溶剂,得到第二次过柱部分;
4)将第二次过柱部分,上样于硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,按石油醚:乙酸乙酯=(5:1)~(1:1)的体积比进行梯度洗脱,对流出液进行检测,将石油醚:乙酸乙酯=3:1的体积比洗脱的馏分合并,蒸干溶剂,得到东莨菪亭。
步骤2)、3)、4)中所用的洗脱液为无毒、低刺激性的石油醚-乙酸乙酯混合溶剂,相比其他常用分离溶剂(如氯仿、甲醇、丙酮、二氯甲烷、乙腈等),本方法所采用的溶剂系统更为环保,且对人体伤害极小。
步骤3)和4)中石油醚-乙酸乙酯系统洗脱液中含有体积分数为0.1~0.4%的甲酸,即石油醚和乙酸乙酯的总体积与甲酸的体积之比均为100:(0.1~0.4)。
本发明所用硅胶的粒度为200~300目。对流出液进行检测是采用薄层色谱法进行检测,于365nm紫外灯下进行检视。
实施例1
从顶花板凳果中提取东莨菪亭的方法,包括以下步骤:
1)将顶花板凳果粗粉约7.5kg,用10倍量的95%乙醇回流提取3次,每次2h,回收提取液,减压浓缩得乙醇提取浸膏,将1.0kg浸膏用10倍量2.0%的盐酸溶解后过滤,滤渣用10倍量的乙酸乙酯研溶后过滤,重复次数为2次,每次2h,将滤液浓缩干燥得乙酸乙酯部位浸膏(32.6g)。
2)取30.0g顶花板凳果乙酸乙酯部位浸膏过常压硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按石油醚与乙酸乙酯体积比为(200:1)~(0:1)的梯度进行洗脱,等量收集洗脱液,每份500ml,收集的馏分用tlc进行检测,于365nm紫外灯下检视,合并相同馏分,蒸干溶剂,共得到27个部分,分别命名为a1-a27,收集洗脱液梯度为石油醚-乙酸乙酯(25:1)~(10:1)的馏分,即a6~a9的馏分,回收溶剂,蒸干,得到第一次过柱部分。其中,所述硅胶粒度为200~300目。
3)将第一次过柱部分过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(25:1)~(1:3)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到第二次过柱部分;
4)将第二次过柱部分再过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(5:1)~(1:1)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到单体化合物85.9mg,通过高效液相色谱法分析纯度在99%以上。
将分离得到的单体化合物采用1h-nmr和13c-nmr等方法进行结构鉴定,并结合文献报道,确定该化合物结构为7-羟基-6-甲氧基香豆素,即东莨菪亭。
核磁共振图谱参照图1和图2,核磁数据如下:淡黄色针状结晶(dmso),分子式c10h8o4,相对分子质量192。
1h-nmr(400mhz,dmso)δppm:10.33(s,1h),7.92(d,j=9.4hz,1h),7.23(s,1h),6.79(s,1h),6.23(d,j=9.4hz,1h),3.82(s,3h)。
13c-nmr(100mhz,dmso)δppm:56.30(och3),103.05(c-8),109.87(c-5),110.86(c-10),112.02(c-3),144.80(c-4),145.52(c-6),149.80(c-9),151.39(c-7),160.99(c-2)。
将本发明得到的东莨菪亭与参考文献中报道的c谱数据进行比对,结果如表1所示:(其中,文献1:wuyb,zhengcj,qinlp,etal.antiosteoporoticactivityofanthraquinonesfrommorindaofficinalisonosteoblastsandosteoclasts[j].molecules,2009,14,573-583.文献2:wuq,zoul,yangxw,etal.antiosteoporoticnovelsesquiterpeneandcoumarinconstituentsfromthewholeherbsofcrossostephiumchinense[j].journalofasiannaturalproductsresearch,2009,11,85-90.)
表1东莨菪亭核磁c谱数据比对
从表1中可以看出,本发明从顶花板凳果中分离得到的单体化合物的c谱数据与文献报道的东莨菪亭的数据基本一致。
实施例2
从顶花板凳果中提取东莨菪亭的方法,包括以下步骤:
1)将顶花板凳果粗粉约7.5kg,用8倍量的95%乙醇回流提取2次,每次3h,回收提取液,减压浓缩得乙醇提取浸膏,将1.0kg浸膏用8倍量1.5%的盐酸溶解后过滤,滤渣用8倍量的乙酸乙酯研溶后过滤,重复次数为3次,每次1h,将滤液浓缩干燥得乙酸乙酯部位浸膏(30.2g)。
2)取28.0g顶花板凳果乙酸乙酯部位浸膏过常压硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,按石油醚与乙酸乙酯体积比为(200:1)~(0:1)的梯度进行洗脱,等量收集洗脱液,每份500ml,收集的馏分用tlc进行检测,于365nm紫外灯下检视,合并相同馏分,蒸干溶剂,收集洗脱液梯度为石油醚-乙酸乙酯(25:1)~(10:1)的馏分,回收溶剂,蒸干,得到第一次过柱部分。其中,所述硅胶粒度为200~300目。
3)将第一次过柱部分过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(25:1)~(1:3)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到第二次过柱部分;
4)将第二次过柱部分再过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(5:1)~(1:1)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到单体化合物,即东莨菪亭,通过高效液相色谱法分析纯度在99%以上。
实施例3
从顶花板凳果中提取东莨菪亭的方法,包括以下步骤:
1)将顶花板凳果粗粉约7.5kg,用12倍量的95%乙醇回流提取4次,每次2.5h,回收提取液,减压浓缩得乙醇提取浸膏,将1.0kg浸膏用12倍量1.0%的盐酸溶解后过滤,滤渣用12倍量的乙酸乙酯研溶后过滤,重复次数为4次,每次1.5h,将滤液浓缩干燥得乙酸乙酯部位浸膏(29.7g)。
2)取28.0g顶花板凳果乙酸乙酯部位浸膏过常压硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,按石油醚与乙酸乙酯体积比为(200:1)~(0:1)的梯度进行洗脱,等量收集洗脱液,每份500ml,收集的馏分用tlc进行检测,于365nm紫外灯下检视,合并相同馏分,蒸干溶剂,收集洗脱液梯度为石油醚-乙酸乙酯(25:1)~(10:1)的馏分,回收溶剂,蒸干,得到第一次过柱部分。其中,所述硅胶粒度为200~300目。
3)将第一次过柱部分过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(25:1)~(1:3)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到第二次过柱部分;
4)将第二次过柱部分再过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(5:1)~(1:1)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到单体化合物,即东莨菪亭,通过高效液相色谱法分析纯度在99%以上。
实施例4
从顶花板凳果中提取东莨菪亭的方法,包括以下步骤:
1)将顶花板凳果粗粉约7.5kg,用15倍量的95%乙醇回流提取2次,每次2h,回收提取液,减压浓缩得乙醇提取浸膏,将1.0kg浸膏用15倍量1.0%的盐酸溶解后过滤,滤渣用15倍量的乙酸乙酯研溶后过滤,重复次数为3次,每次1.5h,将滤液浓缩干燥得乙酸乙酯部位浸膏(27.8g)。
2)取25.0g顶花板凳果乙酸乙酯部位浸膏过常压硅胶柱,以石油醚-乙酸乙酯系统为洗脱液,按石油醚与乙酸乙酯体积比为(200:1)~(0:1)的梯度进行洗脱,等量收集洗脱液,每份500ml,收集的馏分用tlc进行检测,于365nm紫外灯下检视,合并相同馏分,蒸干溶剂,收集洗脱液梯度为石油醚-乙酸乙酯(25:1)~(10:1)的馏分,回收溶剂,蒸干,得到第一次过柱部分。其中,所述硅胶粒度为200~300目。
3)将第一次过柱部分过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(25:1)~(1:3)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到第二次过柱部分;
4)将第二次过柱部分再过硅胶柱,以含0.1%甲酸的石油醚-乙酸乙酯系统为洗脱液,按体积比(5:1)~(1:1)进行梯度洗脱,对流出液用tlc进行检测,于365nm紫外灯下检视,合并主斑点相同的馏分,收集365nm紫外灯下呈现蓝色亮荧光的部分,得到单体化合物,即东莨菪亭,通过高效液相色谱法分析纯度在99%以上。
对比例1
步骤3)和步骤4)中的洗脱液均采用石油醚-乙酸乙酯系统,其它条件同实施例1。
经测试发现,若体系中没有加入甲酸,会导致目标化合物在色谱柱中拖尾并与其他化合物交叉,影响分离度。
对比例2
步骤3)和步骤4)的洗脱液中甲酸的体积浓度依次调节为2%、4%、5%、6%和8%,其它条件同实施例1。
经测试发现,甲酸含量超过0.4%,则会影响洗脱液极性,导致目标化合物过早地被洗脱出,不能成功与其他极性相近的化合物分离。因此,本发明的洗脱液中甲酸含量最多不能超过洗脱液总体积的0.4%。
对比例3
将甲酸替换成有机酸冰醋酸,其它条件同实施例1。
发现常用的有机酸冰醋酸虽然也可达到改善分离效果的目的,但因为其沸点较甲酸更高,会增加后续蒸干浓缩的时间及难度,故不采用冰醋酸。
对比例4
将甲酸替换成碱性溶剂,其它条件同实施例1。
发现产物发生变化,这是因为东莨菪亭中的α-吡喃酮环结构具有α,β-不饱和内酯的性质,在稀碱性溶液中会发生水解,生成不稳定的顺式邻羟基异阿魏酸盐,后者经酸化即闭环恢复为内酯;但若长时间处在碱性环境中或经紫外光照射,则会转变为稳定的反式邻羟基异阿魏酸。故在东莨菪亭的提取分离过程中,不得有碱性物质加入。而甲酸的加入,也会防止东莨菪亭开环分解。东莨菪亭在酸、碱性溶液中的转化过程见下反应方程式:
对比例5
步骤1)中的溶剂乙酸乙酯替换为甲醇,其它条件同实施例1。
发现无法在后续提取液及洗脱液中发现目标化合物东莨菪亭,故使用甲醇提取时无法提取得到该化合物。
通过以上实施例和对比例可知,本发明使用有机酸甲酸加入到洗脱液中可以显著改善拖尾,使化合物更好的与其他成分分离,同时流动相体系中始终不得加入任何碱性溶剂,避免产物分解和异构化,其用量也不能超过4%。
- 还没有人留言评论。精彩留言会获得点赞!