乙胺嗪晶型α及其制备方法与流程
本发明涉及乙胺嗪晶型α及其制备方法。
背景技术:
乙胺嗪即1-二乙胺基甲酰基-4-甲基哌嗪,是合成抗寄生虫病药——枸橼酸乙胺嗪的关键中间体。
de923967c公开了乙胺嗪的结晶方法,用低沸点石油醚作为重结晶溶剂,乙胺嗪熔点为49-50℃,收率为62%。
su1031963a1公开了乙胺嗪的结晶方法,使用丙酮作为重结晶溶剂,乙胺嗪熔点约为51-52℃。
目前,现有技术制备得到的乙胺嗪产品收率及纯度较低,稳定性较差。
因此,现在急需开发出一种收率和纯度更高,稳定性更好的乙胺嗪晶体。
技术实现要素:
为了解决上述技术问题,本发明提供了乙胺嗪晶型α及其制备方法。
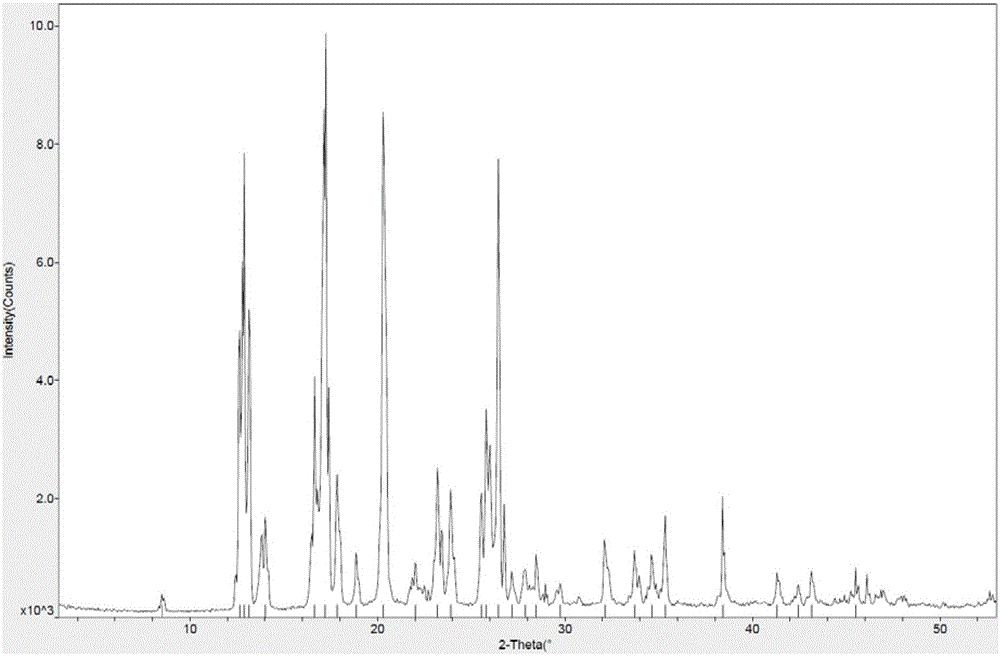
本发明首先提供了乙胺嗪晶型α,所述晶型的x射线粉末衍射中,2θ衍射角度在8.5±0.2°、12.7±0.2°、12.9±0.2°、13.2±0.2°、16.7±0.2°、17.2±0.2°、20.3±0.2°、25.8±0.2°、26.4±0.2°处有特征峰。
进一步地,所述x射线粉末衍射中,2θ衍射角度还在14.0±0.2°、17.9±0.2°、18.9±0.2°、22.0±0.2°、23.2±0.2°、23.9±0.2°、25.5±0.2°、28.5±0.2°处有特征峰。
进一步地,该晶型的x射线粉末衍射中,2θ衍射角度特征峰的相对强度值为:
进一步地,该晶型的熔点为49-50℃。
本发明还提供了制备乙胺嗪晶型α的方法,它包括以下步骤:
(1)将乙胺嗪溶解于有机溶剂中,所述有机溶剂不选自石油醚或丙酮中的任一种或其组合;
(2)于-20~20℃结晶;
(3)分离出晶体,干燥即得。
进一步地,步骤(1)中的溶解过程中,温度为10℃至溶剂的回流温度。
进一步地,步骤(1)中,所述溶剂选自正戊烷、正己烷、正庚烷、环戊烷、环己烷、环庚烷、丙醚、甲乙醚、异丙醚、甲基异丙基醚、甲基叔丁基醚、四氢呋喃、2-甲基四氢呋喃、四氢吡喃、甲基异丙基酮、甲基异丁基酮、1,4-二氧六环、甲酸乙酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸丁酯、乙酸异丁酯、乙酸叔丁酯、乙腈中的任一种或其组合;
进一步地,所述溶剂与乙胺嗪的体积重量比为0.5~5ml/g。
试验结果证明,本发明制备得到的乙胺嗪晶型α比现有的乙胺嗪晶型具有更高的纯度以及更好的稳定性。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
图1为本发明乙胺嗪晶型α的xrpd图。
图2为对比例1中乙胺嗪对照晶型的xrpd图。
具体实施方式
实施例1、本发明晶型α的制备
取10.0g乙胺嗪粗品加入到50ml环己烷中,加热搅拌溶清。降温至-20℃析晶。过滤,固体于35℃真空干燥,得到8.4g乙胺嗪晶型α。收率84.0%,化学纯度99.98%,xrpd图谱如图1所示,xrpd数据如表1所示。
表1本发明乙胺嗪晶型α的xrpd数据
实施例2、本发明晶型α的制备
取15.5g乙胺嗪粗品加入8ml甲基叔丁基醚中,加热搅拌溶清。降温至-10℃,析晶。过滤,固体于30℃真空干燥,得到12.8g乙胺嗪晶型α。收率82.6%,化学纯度99.96%。
实施例3、本发明晶型α的制备
取20.4g乙胺嗪粗品加入到50ml甲基异丙基酮中,加热搅拌溶清。降温至10℃,析晶。过滤,固体于25℃真空干燥,得到16.4g乙胺嗪晶型α。收率80.4%,化学纯度99.97%。
实施例4、本发明晶型α的制备
取27.6g乙胺嗪粗品加入到30ml乙酸乙酯中,加热搅拌溶清。降温至0℃,析晶。过滤,固体于20℃真空干燥,得到22.6g乙胺嗪晶型α。收率81.9%,化学纯度99.99%。
以下通过试验例的方式来具体说明本发明的有益效果。
对比例1
取将10.0g乙胺嗪粗品用石油醚重结晶,得到6.7g乙胺嗪对照晶型,熔点48-50℃。收率67.0%,化学纯度99.55%(参考de923967c),其xrpd图谱如图2所示,xrpd数据如表2所示。
表2乙胺嗪对照晶型的xrpd数据
试验例1、晶型的稳定性评价
1)实验材料
实施例4制备的乙胺嗪晶型α;对比例制备的乙胺嗪晶型。
2)实验方法与结果
试验方法参见《中国药典(2015)》第四部9001《原料药物与制剂稳定性试验指导原则》。
影响因素试验:
①高温试验:取乙胺嗪对照晶型以及实施例4乙胺嗪晶型α,于40℃温度下放置10天,于第5天和第10天取样,测定各项指标与0天样品进行比较(hplc法),试验结果见表3。
②强光照射试验:取乙胺嗪对照晶型以及实施例4乙胺嗪晶型α,于照度为(4500±500)lx的条件下放置10天,于第5天和第10天取样,测定各项指标与0天样品进行比较(hplc法),试验结果见表3。
表3乙胺嗪晶型影响因素试验结果
加速试验:
将制得的乙胺嗪对照晶型以及实施例4乙胺嗪晶型α在恒温恒湿箱中进行6个月的加速试验。试验条件是:(25±2)℃/rh(60±5)%,分别于0、1、2、3、6个月取样,进行化学纯度和杂质检验(hplc法),结果见表4。
表4乙胺嗪晶型加速试验结果
从表3和表4可知,本发明乙胺嗪晶型α的稳定性比对照晶型好,特别是在光照条件下,本发明乙胺嗪晶型α的稳定性与对照晶型相比有明显的提高。
综上,本发明制备得到了纯度更高,稳定性更好的乙胺嗪新晶型。
- 还没有人留言评论。精彩留言会获得点赞!