NGS文库浓度的标准化的制作方法
本申请要求2016年9月6日提交的美国临时专利申请号62/384,118的优先权,其通过引用全文纳入本文。
背景技术:
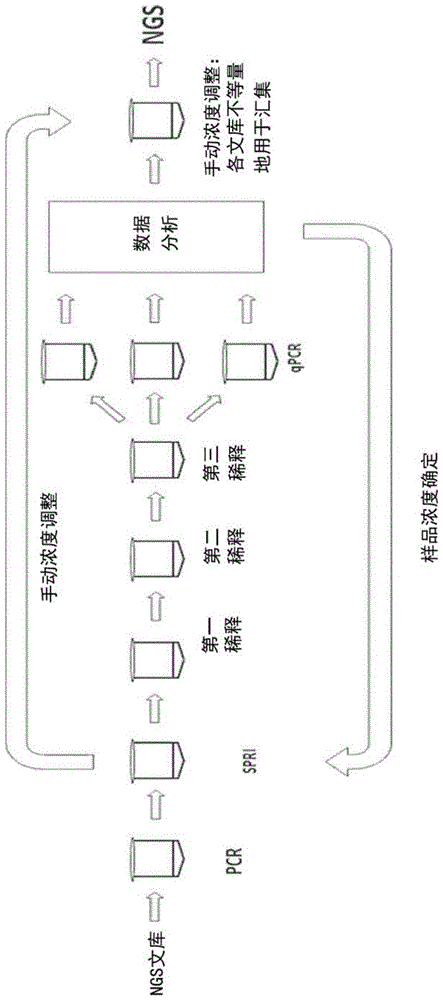
:为了使高质量下一代测序(ngs)数据最大化,至关重要的是给测序仪器加载数量准确的文库dna。文库dna装载数量不足会导致簇密度低(illumina)/模板阳性isp低(iontorrent)并且降低测序输出,而文库dna过剩会导致嵌合簇(illumina)/多克隆isp(iontorrent)并导致低量低质量数据。当汇集文库用于并行测序时,不准确的定量会导致测序数据不平衡,其中非足量文库被过度测序,而过量文库则测序不足。出于这些原因,可测序文库分子数量的准确定量在ngs工作流程是至关重要的一步。当汇集文库以产生用于杂交捕获的并行集合时,同样需要准确的文库定量。当前用于文库定量的方法包括芯片电泳(例如,安捷伦生物分析仪(agilentbioanalyzer),dsdna的荧光测定方法(例如,qubit)和qpcr(各种市售可得的试剂盒)。芯片电泳和荧光测定方法两者都仅能准确定量pcr扩增的、对全部加了衔接子的文库分子进行了富集的文库,因为这些方法不能区分无pcr文库制备物中的功能性分子(包含两个ngs衔接子的片段)和非功能性分子(仅包含1种或没有ngs衔接子的片段)。qpcr在ngs工作流程中被广泛地使用,因为其能够准确定量功能性文库分子,但是该方案涉及多次移液步骤并且花费大量的时间。各文库必需经连续稀释,qpcr试验按照标准曲线一式三份运行,然后进行qpcr数据分析。qpcr定量过程接近2个小时,包括至少45分钟操作时间。最终,当汇集样品时,所有这些方法都需要针对各ngs文库手动浓度调整。此外,当前所有文库定量都依赖于文库插入大小(insertsize)将质量转化成摩尔浓度,并且,如果文库的尺寸分布宽或尺寸不确定,那就无法准确确定摩尔定量。技术实现要素:本公开提供了不依赖于文库插入大小的用于ngs文库摩尔标准化的新方法。相对于文库定量然后手动调整文库浓度(图1a),该方法提供了简便的替代方法。本文所述文库标准化在单个步骤中或多个步骤中发生,以产生处理等摩尔ngs文库量的数个样品。需要单个步骤的方法是基于pcr的标准化(n-pcr),其中使用限定浓度的标准化引物(n-pcr引物)以加快的退火动力学和达成pcr反应期间引物全利用的扩增条件进行各ngs文库的扩增,由此提供指定摩尔量的各ngs文库。需要多个步骤的方法是酶法标准化,这需要:a.用预标准化(pre-normalization)引物(pre-n引物)pcr扩增得过量的各ngs文库,所述预标准化引物在pcr中或pcr后的酶促消化过程中在一端或两端产生5'或3'突出端,然后进行纯化步骤;b.各扩增所得文库与指定摩尔量的标准化探针(n-探针(n-probe))-有些实施方式中还有dna连接酶-孵育,其中探针退火并连接到5'或3'突出端从而选出与n-探针等摩尔的文库部分;和c.对于一些实施方式,通过酶促消化未获选文库部分或通过从固相固定化中酶介导释放探针选定部分进行探针选定部分的分离。酶法标准化方法的示例性工作流程如图1b所示。可以看出的是,相比如图1a所示的qpcr定量和手动调整,该过程可以减少人工操作和总时间。因此,通过限制标准化pcr引物浓度或通过限制n-探针浓度,实现了文库标准化(参见例如图2所示的模拟文库标准化,虚线表示标准化探针的量)。如图2所示,可以使用本公开的方法标准化文库,从而可以消除过量的文库,产生等摩尔的文库浓度。本文所述ngs文库标准化快速、无需连续稀释或手动浓度调整,因此与qpcr定量相比节省多达1小时的操作时间和多达2小时的总准备时间。该方法可以在加载测序仪器前用于ngs文库摩尔调节或在杂交捕获之前用于文库汇集(分别如图3a和3b所示)。其与使用illumina、iontorrent或对pcr扩增的dna文库进行测序的其他平台的任何ngs文库(dna或rna)相兼容。本文所述可选方案并不利用限定链霉亲和素珠结合步骤,该步骤需要文库输入高度过量且导致回收一致性(recoveryconsistency)差,这导致摩尔浓度输出范围不确定。此外,本文所述方法是简单的,孵育的移液步骤很少,易于自动化。本文所述方法需要通过pcr进行文库扩增并且不可以直接用于无pcr文库方案,但可以经调整后与无pcrngs文库联用。此外,对于酶法标准化,pcr后得率低于指定摩尔阈值的文库在标准化过程后将保持偏低摩尔量,从而导致所得文库代表数不足,测序或杂交捕获工作流程中数据输出偏低(图2,样品3)。为此,对于每个文库都建议生成相对于较少摩尔量n-探针摩尔量过量的文库,从而避免产生代表数不足的样品。一些本文所述标准化方法需要这样的pcr扩增步骤:它产生标准化的扩增文库(在n-pcr的情况中)或者产生位于一个或两个衔接子末端的5'或3'突出端(在酶法标准化的情况中)。在一些示例中,突出端并不是必需的。突出端形成于pcr过程中或pcr后的酶促消化期间,从而使n-探针能够退火结合dsdna底物。酶促标准化方法中有限的、指定摩尔量的n-探针退火结合等摩尔量的ngs文库分子(一些情形中是通过双链ngs文库的5'或3'突出端)。在一些情形中,不需要突出端,探针能够通过例如但不限于以下所述的方法与扩增所得文库中的文库分子连接:钝端连接,ta连接和粘性末端连接。因此,精确的n-探针量选定酶法标准化过程回收的文库分子数量。基于pcr的标准化具有有限的、指定摩尔量的n-pcr引物,所述n-pcr引物扩增等摩尔量的ngs文库。因此,精确的n-pcr引物量选定n-pcr过程生成的文库分子数量。基于pcr的标准化当使用常规试剂试图通过限制引物浓度来控制pcr产物的量时,多个因素素降低了这种方法的实用性。首先,常规pcr引物范围在200nm至大于1um,完全利用如此高浓度的引物将产生10-50pmol的pcr产物,这需要高浓度的dna聚合酶并且造成样品的严重过度扩增,而这是不利的因为会引入复制错误和碱基组成偏性,这在扩增ngs文库时特别重要。或者,可以较低的20-40nm引物浓度进行pcr产生1-2pmol的扩增产物,但是在这样的情况下,引物退火时间将需要相应地增加10倍以保证有效的引物退火、延伸和无偏性扩增高复杂性模板(如ngs文库),其中过多的热循环孵育时间也是不利的,因为可能造成dna损伤并且降低ngs文库质量。在本文所述某些实施方式中,引入了解决这些问题的新型标准化pcr引物组合物(n-pcr引物)。该标准化pcr引物组合物采用常规退火时间的扩增循环,提高了引物退火杂交速率,减少了退火时间并且能够有效并完全利用pcr引物,由此通过限制pcr引物浓度可再现地生成指定摩尔量的ngs文库。为了提高引物杂交速率,将包含低复杂性序列的5'尾导入引物对的各n-pcr引物,各引物的3'部分退火结合已经存在于文库模板上的ngs衔接子序列。在一些实施方式中,正向和反向n-pcr引物具有不同的5'尾序列。在另一些实施方式中,正向和反向n-pcr引物具有相同的5'尾序列。低复杂性5'尾可以包含均聚物重复序列,如(a)n、(t)n、(g)n或(c)n,二核苷酸重复序列,如(ag)n、(ac)n、(gt)n、(ct)n、(at)n或(gc)n,三核苷酸,四核苷酸,五核苷酸或甚至更大的重复序列元件。n-pcr引物的5'尾长度可以是8至50个碱基或更多,包含具有或不具有其他修饰的脱氧核苷酸或核糖核苷酸,或者可以是它们的混合物。在一些实施方式中,当n-pcr引物扩增在各末端包含独特衔接子序列的ngs文库时,n-pcr引物的3'部分退火结合的衔接子序列针对正向和反向引物是不同的,然而在另一些实施方式中,当正向和反向n-pcr引物扩增文库的两个末端衔接子相同的ngs文库时,正向和反向n-pcr引物退火结合相同的衔接子序列。在前两个循环期间,当使用限定浓度的n-pcr时,引物退火结合模板仅通过与ngs衔接子互补的n-pcr引物3'部分发生。由此,应当延长最初这些循环的退火时间从而确保低引物浓度时所有文库分子的延伸。一旦低复杂性/重复性尾序列的反向互补链纳入扩增子后,n-pcr引物的5'和3'部分都会参与退火结合模板,并由于低复杂性组成显著地使退火加快,后续循环因此可采用常规退火时间,由此能够实现文库的有效pcr扩增。由于低复杂性/重复性5'尾序列先快速退火然后高复杂性3'衔接子序列退火,因此引物杂交加快。待完成n-pcr引物的完全利用即生成了指定摩尔量的ngs文库。由于有限的引物浓度,所得扩增文库可能主要是单链的,但是其中会发生衔接子序列的再退火从而生成部分双链的异源双链核酸分子。任选地,如果需要,在进行测序之前,可以从文库切掉低复杂性尾序列。或者,可以在文库制备的衔接子连接步骤中将与各n-pcr引物5'尾互补的低复杂性序列结合在ngs衔接子末端由此将其引入。在该实施方式中,在n-pcr扩增的每个循环周期都可以采用常规退火时间,因为低复杂性序列在pcr前已经存在于完成的ngs文库底物上。酶法标准化在该方法中,将使用预标准化引物(pre-n引物)的pcr扩增用于产生相对于标准化探针(n-探针)量过量的摩尔量,所述n-探针将被用于选择文库分量。n-探针可用于多种方法(图4描述了示例性的方法)。在一实施方式中,pre-npcr引物具有生物素基团和至少一个rna碱基,并且使用链霉亲和素珠捕获该生物素标记的整个ngs文库,其中仅具有退火n-探针的选定摩尔分量从珠固定中释放,因为该探针与所述rna碱基重叠形成了被rna酶h酶促切割的底物(方法1;受控的释放)。在其他实施方式中,n-探针与选定文库部分的退火和连接赋予该文库部分针对酶促消化的核酸酶抗性(方法2;受控的保护)。在这些可选方案中,n-探针包含赋予酶促消化抗性的碱基、基团或构象。在其他实施方式中,n-探针与选定文库部分退火和连接发生在酶促消化以及去除特异性、功能性文库序列之后,其中,n-探针连接导致选定文库部分的文库功能性恢复(方法3;受控的修复)。在另一实施方式中,n-探针连接完成文库合成,其中仅指定的与探针连接的摩尔分量生成功能性文库分子(方法4;受控的合成)。方法1需要3个标准化步骤,方法2需要2步,而方法3和4有一步,都是pcr后的。没有任何限制,应当理解的是,这些不同方法的各个方面可以任意组合使用以实现ngs文库的摩尔标准化。为了能够使n-探针退火结合dsdna文库底物,存在至少3种方式可以生成5'突出端用于标准化步骤。在一些实施方式中,在pcr期间使用预标准化引物(pre-n引物)在一个或两个文库末端产生5'突出端,所述pre-n引物具有用于n-探针退火的5'尾以及位于尾和衔接子序列之间的不可复制间隔区,该不可复制基团(group)包括但不限于du碱基(针对古细菌(archael)dna聚合酶),稳定的脱碱基位点,如d间隔子(dspacer),r间隔子(rspacer),间隔子c3、c6或c12,己二醇,三乙烯乙二醇间隔子9(spacer9)和六乙烯乙二醇间隔子18(spacer18)。在其他实施方式中,在pcr期间使用具有3'外切核酸酶校对活性的热稳定dna聚合酶和pre-n引物在一个或两个文库末端产生5'突出端,所述pre-n引物具有用于n-探针退火的5'尾以及新型不可复制间隔区,其包含位于尾和衔接子序列之间的3个或更多核糖u(ribou)或核糖a(riboa)碱基的连续串,其中高保真dna聚合酶不能够通过(核糖u)n或(核糖a)n模板延伸,其中n=3或更多。本文所述(核糖u)n/(核糖a)n复制挡块的独特之处在于其可以通过非校对聚合酶复制,如taqdna聚合酶,并且还能够连接与5'尾和多聚(ru)或多聚(ra)串互补的探针寡核苷酸,不同于生成不可连接的转接区的其他复制封闭基团。在另一些实施方式中,pcr后使用t4dna聚合酶产生位于一个或两个文库末端的5'突出端。在该情况中,pre-npcr引物纳入5'尾和3'侧缓冲区。尾区是10-20个碱基并且包含均聚物、二-或三-核苷酸组合,后跟5-10个碱基的缓冲区,该缓冲区具有5'尾区所没有的核苷酸组成。当用t4dna聚合酶以及限制为仅与缓冲区而非尾区碱基互补的核苷酸混合物孵育包含这类序列的ngs文库时,t4dna聚合酶的3'外切核酸酶校对活性将会不可逆地修整3'互补尾区直到其达到缓冲区,而在此,其能够可逆地去除并替换核苷酸,由此产生据缓冲区限定的5'突出端。或者,存在至少2种不同的方式可以生成3'突出端用于n-探针退火。在一些实施方式中,在pcr后,通过用诸如rna酶h等酶、udg和脱碱基内切核酸酶或内切核酸酶v的混合物孵育,通过切割由包含那些可切割碱基的经修饰的pre-npcr引物纳入的rna,脱氧尿嘧啶或脱氧肌苷碱基,产生位于一个或两个文库末端的3'突出端。在其他实施方式中,在pcr后,通过用5'外切核酸酶诸如t7外切核酸酶或λ外切核酸酶孵育产生位于一个或两个文库末端的3'突出端。在这些实施方式中,3'突出端的长度由pre-npcr引物内可切割/核酸酶抗性碱基或连接和整体引物长度控制。在一些实施方式中,n-探针连接ngs的3'或5'端,其中dna连接酶是t4dna连接酶,t3dna连接酶,t7dna连接酶,或大肠杆菌dna连接酶,热稳定dna连接酶,如taq连接酶,amp连接酶,9°ndna连接酶或pfudna连接酶。在一些实施方式中,不进行n-探针连接。在一些其他实施方式中,探针连接还可以包括置换和切割用rna酶h切割后残留的5'rna碱基、或用udg和脱碱基内切核酸酶或内切核酸酶v的混合物不完全消化残留的脱氧尿嘧啶或脱氧肌苷修饰碱基、或t7外切核酸酶或λ外切核酸酶消化后残留的核酸酶抗性碱基。在这样的情况中,连接反应中可以补充taqdna聚合酶或dna聚合酶i以及(如果有必要)限定核苷酸混合物,以允许有限的断口(nick)平移反应。一些实施方式在n-探针连接后还需要分离探针选定文库的步骤。对于方法1,用于探针选定文库切割的酶(由固定释放)包括rna酶h酶,包括大肠杆菌rna酶h1和rna酶h2,或热稳定rna酶h。在其他实施方式中(方法2),用于非探针选定文库消化的酶包括3'外切核酸酶,如外切核酸酶iii,t4dna聚合酶,外切核酸酶i,以及5'外切核酸酶,如外切核酸酶t7或λ外切核酸酶。没有任何限制,应当理解的是,用于5'和3'突出端产生、n-探针连接和文库部分富集或消化不同方法的各个方面可以任意组合使用以实现ngs文库的摩尔标准化。附图说明当结合附图进行阅读时,将更好地理解前面的
发明内容以及下面的示例性实施方式的详细说明。为了说明,附图中显示本发明的示例性构建。然而,本发明并不局限于本文所述的具体方法和手段。在以下附图中,p5和p7分别用于指p5和p7ngs衔接子。图1a和1b比较了qpcr文库定量(图1a)与通过本文所述酶法处理的标准化反应(图1b)。图2显示本文所述标准化方法之一的结果,该方法产生等摩尔浓度的各样品的结果,无需手动调整各个样品的量。图3a显示本文所述标准化方法的示例性工作流程,据此,文库制备后,对文库进行pcr,然后通过spri纯化并进行标准化以产生标准化的样品,用于ngs。图3b显示包含有标准化过程的示例性并行工作流程。图4显示本公开用于标准化的4种示例性方法,其中,第一步涉及ngs文库的pcr以及通过spri纯化扩增的ngs文库,然后是标准化探针退火和任选的连接,然后是富集探针选定部分。图5显示通过方法1(受控的释放)的酶法标准化的示例性方法。“b”表示生物素。图6显示通过方法1(受控的释放)的酶法标准化的示例性方法。“b”表示生物素。红色条指示rna碱基的部分。黑色条指示不可复制的dna间隔区的位置。图7a显示通过方法2(a)(受控的保护)和方法2(b)(受控的保护)的酶法标准化的示例性方法。图7b显示通过方法2(c)(受控的保护)的酶法标准化的示例性方法。图8a显示通过方法2(a)使用1个pre-npcr引物靶向1个文库链的酶法标准化的示例性方法。图8b显示通过方法2(a)靶向1个文库链的酶法标准化的示例性方法,产生单链dna文库。图8c显示通过方法2(a)使用2个pre-npcr引物靶向两个文库链的酶法标准化的示例性方法。图8d显示通过方法2(a)使用1个pre-npcr引物靶向两个文库链的酶法标准化的示例性方法,所述1个pre-npcr引物在文库中产生单链dna和双链dna。图9a显示使用pre-npcr引物和常规引物靶向1个文库链的用于方法2(a)的pcr扩增的示例性方法。图9b显示使用pre-npcr引物和常规引物靶向1个文库链的用于方法2(a)的pcr扩增的示例性方法,其具有使用标引引物的先期pcr步骤以完成具有截短衔接子的ngs文库。图10a显示使用2个pre-npcr引物靶向两个文库链的用于方法2(a)的pcr扩增的示例性方法。图10b显示使用2个pre-npcr引物靶向两个文库链的用于方法2(a)的pcr扩增的示例性方法,其具有使用标引引物的先期pcr步骤以完成具有截短衔接子的ngs文库。图11显示用于使用t4dna聚合酶通过方法2(b)(受控的保护)以及消化生成5'突出端的示例性方法,用于ngs文库标准化。图12显示用于生成3'突出端的示例性方法,使用5'外切核酸酶,用于方法2(c)(受控的保护)。图13显示通过方法3(a)、3(b)和3(c)的受制ngs衔接子修复进行的酶法标准化的示例性方法。图14显示使用rna酶h、连接酶和外切核酸酶i通过方法3a(受控的修复)进行标准化的示例性方法。图15显示使用rna酶h和连接酶通过方法3b(受控的修复)进行标准化的示例性方法。图16显示使用5'外切核酸酶和连接酶通过方法3c(受控的修复)进行标准化的示例性方法。图17显示通过方法5(a)和5(b)的受控pcr后文库合成(受控的合成)进行酶法标准化的示例性方法。图18显示使用方法4(受控的合成)靶向1个ngs文库链的通过合成进行标准化的示例性方法。图19显示使用方法4(受控的合成)靶向两个ngs文库链的通过合成进行标准化的示例性方法。图20a显示使用p5末端标准化单标引(以带标引的探针进行双重标引)靶向1个文库链的通过方法4标准化的pcr扩增的示例性方法。图20b显示使用p7末端标准化、双重标引靶向1个文库链的通过方法4标准化的pcr扩增的示例性方法。图21a显示靶向ngs文库双链的通过方法4(受控的合成)标准化的pcr扩增的示例性方法。图21b显示方法4(受控的合成)使用标准化探针用于单标引或双重标引的示例性方法。图22显示使用退火速率加快的n-pcr引物用于通过受控的扩增进行标准化的示例性方法。图23a显示通过受控的扩增使用n-pcr引物二核苷酸重复5'尾区基于pcr标准化的示例性方法。图23b显示通过受控的扩增使用n-pcr引物均聚物重复5'尾区基于pcr标准化的示例性方法。图24a显示n-pcr期间的文库浓度和n-pcr引物。图24b显示n-pcr期间的文库浓度和n-pcr引物。图24c显示n-pcr期间的文库浓度和n-pcr引物。图24d显示n-pcr期间的文库浓度和n-pcr引物。图25显示具有均聚物尾、二核苷酸尾或三核苷酸尾中任一个的示例性n-pcr引物。图26显示文中提出的n-pcr引物加快退火的机制。图27显示使用n-pcr引物扩增和标准化ngs文库的示例性方法。图28显示使用n-pcr引物产生预订量pcr产物的pcr的示例性方法。图29a显示文中提出的当两个n-pcr引物具有相同的尾序列时形成二级结构的机制。图29b显示文中提出的当n-pcr引物具有互补的尾序列时n-pcr引物之间干扰的机制。图29c显示文中提出的当使用具有非互补均聚物尾的n-pcr引物时由于形成非沃森-克里克二级结构导致复制失败的机制。图29d显示具有二核苷酸重复尾序列的示例性n-pcr引物。图30显示pcr后ngs文库摩尔浓度定量的示例性方法。图31显示在含有核糖核苷酸复制阻断子的dna模板上的引物延伸反应示意图。图31b显示图31a引物延伸反应的预期凝胶电泳结果。图32显示用sybr金染色的15%tbe-urea凝胶上实施例2中延伸反应的产物。“*”指示模板;“**”指示完全延伸的引物;和“***”指示部分延伸的引物。图33a显示用sybr金染色的15%tbe-urea凝胶上实施例3中采用taq和q5du绕过的延伸反应的产物。“*”指示模板;“**”指示完全延伸的引物;和“***”指示部分原始的引物。图33b显示用sybr金染色的15%tbe-urea凝胶上实施例3中采用κhifi或gxl的延伸反应的产物。“*”指示模板;“**”指示完全延伸的引物;和“***”指示部分原始的引物。图33c显示用sybr金染色的15%tbe-urea凝胶上实施例3中采用q5聚合酶的延伸反应的产物。“*”指示模板;“**”指示完全延伸的引物;和“***”指示部分原始的引物。图34a显示采用标准扩增引物和含有复制阻断子和5'端非互补尾序列的引物的扩增实验的示意图。图34b显示用sybr金染色的15%tbe-urea凝胶上实施例5中采用taq和gxl的延伸反应的产物。“**”指示全长扩增产物,而“***”指示截短的扩增产物。图35a显示荧光标记的探针与淬灭剂寡核苷酸之间结合的示意图。图35b显示25nm荧光复杂序列探针与过量的具有淬灭剂的互补底物寡核苷酸(虚线)结合和25nm荧光均聚序列探针与具有淬灭剂的其互补底物(实线)结合的荧光强度的时间曲线。图36a显示实施例7中文库1-7的预标准化前浓度。图36b显示实施例7中文库1-7的标准化后最终浓度。图37a显示荧光探针与过量的具有淬灭剂的两种不同底物寡核苷酸结合的荧光强度的时间曲线。图37b显示荧光探针与具有淬灭剂的常规模板结合的示意图。图37c显示荧光探针与具有淬灭剂的gr加尾模板结合的示意图。图38a显示实施例9中常规引物联用的文库1-7的文库输入浓度。图38b显示实施例9中使用常规引物进行pcr后文库1-7的产率。图38c显示实施例9中与n-pcr引物联用的文库1-7的文库输入浓度。图38d显示实施例9中使用n-pcr引物进行n-pcr后文库1-7的产率。图39a显示实施例10中文库1-8的文库输入浓度。图39b显示使用n-pcr引物进行n-pcr后文库1-8的产率。图39c显示使用n-pcr标准化的文库进行下一代测序时文库1-8在流动池上形的成簇%。图40a显示实施例11中文库1-16的文库输入浓度。图40b显示实施例11中文库1-16经过标准化的文库浓度。图40c显示实施例11在添加淬灭剂前的相对文库浓度。图40d显示实施例11在添加淬灭剂后的相对文库浓度。图41a显示本公开示例性标准化方法的示意图。图41b显示本公开示例性标准化方法的示意图。图41c显示本公开示例性标准化方法的示意图。图41d显示本公开示例性标准化方法的示意图。图41e显示本公开示例性标准化方法的示意图。图42a显示本公开示例性标准化方法的示意图。图42b显示本公开示例性标准化方法的示意图。图42c显示本公开示例性标准化方法的示意图。图41d显示本公开示例性标准化方法的示意图。图41e显示本公开示例性标准化方法的示意图。图41f显示本公开示例性标准化方法的示意图。图41g显示本公开示例性标准化方法的示意图。图42h显示本公开示例性标准化方法的示意图。图42i显示本公开示例性标准化方法的示意图。图43a显示实施例12。图43b显示实施例12。图43c显示实施例12。图43d显示实施例12。图44显示实施例13。图45a显示实施例14图45b显示实施例14。图45c显示实施例14。图46a显示实施例14。图46b显示实施例14。图47a显示实施例14。图47b显示实施例14。图48a显示实施例15。图48b显示实施例15。图48c显示实施例15。图48d显示实施例15。图48e显示实施例15。图49a显示实施例16。图49b显示实施例16。图49c显示实施例16。图50a显示实施例17。图50b显示实施例17。说明本公开参照某些附图描述了特定的实施方式,但是主题不限于此。描述的附图仅是说明性的且是非限制性的。在附图中,为说明的目的,一些元素的尺寸可能被夸大或扭曲且未按比例绘制。除非特别指明,本文所述元件在首次出现时用“一(个)”或“一(种)”表示,在第二次或后续出现时用“该”或“所述”表示。本公开将提供对附图的描述,其中显示的只是本文所述主题的部分但非全部实施方式。实际上,主题可以以多种不同的形式实施,不应认为仅限于本发明所述的实施方式;而是,提供这些实施方式是为了使本公开内容满足所有法律要求。以下描述中所使用的某些术语仅是为了方便而不是限制。本文所用某些词语指定了在相关附图中的方向。除非特别指明,文中术语“一(个)”、“一(种)”、和“该/所述”并不限于不限于一个要素,而应该被理解为意思是“至少一个”。本文中所用的“另一(种/个)”表示至少有第二种/个或更多种/个。术语包括上述词语,其衍生词以及类似含义的词语。在权利要求中使用术语“或”表示“和/或”,除非特别指出仅表示彼此替换或替换选项彼此排斥。术语“约”用于数值旨在包括+/-10%。例如,如果氨基酸数量表示约200,那么这包括180至220(正或负10%)。除非另外定义,本文中所使用的所有技术和科学术语具有本领域普通技术人员通常所理解的同样含义。酶法文库标准化方法方法1:通过从固定化中受控释放进行酶法文库标准化该方法利用由磁珠固定中酶促释放指定摩尔量的文库。该方法包括至少3个步骤:pre-npcr步骤后纯化,然后是捕获步骤和受控的洗脱步骤。没有任何限制,应当理解的是,该方法的各个方面和不同实施方式可以任意组合使用以实现ngs文库的摩尔标准化。图5显示了该方法示例性的4步骤形式,其包括如下步骤:(1)使用具有不可复制间隔区的引物进行pcr扩增,从而在ngs衔接子一端导入具有位于5'端的生物素和rna切割位点的5'突出端的,并进行spri纯化;(2)通过指定量的标准化探针退火结合5'突出端来选定指定数量的文库分子;(3)珠捕获整个文库(步骤2和3可以在同一反应中进行或以先2后3或先3后2的顺序进行);和(4)通过与rna酶h孵育(在rna碱基处切割),从珠上释放含有标准化探针的文库的指定摩尔分量。该方法需要pcr扩增相对于将使用的n-探针量摩尔过量的ngs文库。为此pcr,pre-n引物在扩增所得ngs文库的一个末端导入5'或3'突出端,并且对于一些实施方式,使用具有3'外切核酸酶校对活性的热稳定性dna聚合酶。可以使用任何本文所述方法生成5'或3'突出端(复制挡块在图6使用的引物中有所显示)。对于该方法,pre-n引物还需要生物素基团和至少一个rna碱基,优选位于所得5'或3'突出端的中间。扩增后,进行纯化步骤,然后进行整个扩增文库的链霉亲和素磁珠捕获,并且该同一捕获反应中,包括限定摩尔量的n-探针,该n-探针与5'或3'突出端互补,优选包含均聚物序列,并且退火结合等摩尔量的固定化的文库,其中,相对于具有复杂序列组成的探针,均聚物序列在探针或底物不显著摩尔过量的情况下促进退火。在最后一步中,通过使用rna酶h的酶促切割来洗脱指定摩尔量的dsdna文库,其中仅有选定的结合n-探针的分量是rna酶h切割的底物因而从固相固定化中被释放,因为探针与包含rna碱基的突出端杂交。方法2:通过受控保护免于外切核酸酶消化进行酶法文库标准化图7a-7b概述了用于进行方法2的三种示例性可选方案,其中在pre-npcr扩增后,方法2a具有连接步骤和消化步骤,而方法2b和c具有切割/连接步骤和消化步骤。在方法2a和b中,生成了5'突出端,而在方法2c中,生成了3'突出端,这些突出端可使用任何本文所述方法产生。所有三种标准化方案需要pcr扩增相较于将使用的n-探针摩尔量过量的ngs文库。如果一个pre-n引物与常规引物一起使用,那么pre-n引物在dsdna文库的一端导入5'或3'突出端,或者如果使用两个pre-n引物,那么pre-n引物在dsdna文库的两端引入5'或3'突出端。在方法2a中,在pcr期间使用pre-n引物生成5'突出端,所述pre-n引物包含3个或更多核糖u或核糖a碱基连续串阻止具有3'外切核酸酶校对活性的热稳定性dna聚合酶进行的复制。在方法2b中,基于标准化反应中pre-n引物和dntp组合物的特定核苷酸组成,通过t4dna聚合酶3'外切核酸酶活性在pcr后生成5'突出端。在方法2c中,在通过酶促切割纳入的引物序列在pcr后生成3'突出端,其中pre-n引物将可切割的碱基导入pcr产物中。对于所有三种方法,pcr扩增后,进行纯化步骤,并且在第一个酶法标准化步骤中,添加限定摩尔量的n-探针和连接酶;对于方法2b和c,还将切割酶添加到该反应中。n-探针与5'或3'突出端互补,包含核酸酶抗性修饰,并且优选包含低复杂性序列,如均聚物,并且退火和连接至等摩尔量的扩增文库。相对于具有复杂序列组成的探针,均聚物序列在探针或底物不显著摩尔过量的情况下促进退火。在第二个酶法标准化步骤中,进行无探针连接的过量文库部分的酶促消化,其中n-探针指定摩尔量的文库保持核酸酶抗性并且被保护免于消化。对于方法2a和b,使用3'外切核酸酶,而对于方法2c,使用5'外切核酸酶。形成5'或3'突出端并非n-探针连接的必要条件,但是突出端显著促进低浓度时探针与文库连接。在一实施方式中,双链标准化探针与单t-碱基3'突出端的连接需要在pcr期间通过taqdna聚合酶产生的具有单a-碱基3'突出端的文库。在另一实施方式中,双链标准化探针具有钝端,并且连接使用高保真dna聚合酶扩增的钝端文库。pcr产生的高文库浓度有助于限定量的钝端或单t-碱基3'突出端标准化探针与文库的截短衔接子末端的连接。预防探针连接至ngs文库相对端的衔接子可以通过使衔接子末端缺少5'磷酸基团或缺少相容的钝端或单a碱基突出端或同时缺少用于探针连接的相容的末端和5'磷酸来控制。具体地,在图7a中,方法2(a)和2(b)包括4个步骤:(1)进行pcr扩增以在一个或两个衔接子末端导入探针结合序列,所用pcr引物含有不可复制的碱基或碱基组合以在pcr期间产生5'突出端(方法2a),或具有特定碱基组成从而能够通过t4dna聚合酶获得5'突出端(方法2b),然后进行spri纯化;(2)通过t4dna聚合酶介导的3'-5'外切核酸酶消化生成5'突出端(仅方法2b);(3)通过指定量的标准化探针退火并连接5'突出端保护指定数量的文库分子;和(4)3'外切酶消化无探针保护的文库部分。具体而言,在图7b中,方法2(c)包括4个步骤:(1)进行pcr扩增以在一个或两个衔接子末端导入探针结合序列,所用pcr引物含有可切割碱基从而能够通过酶促切割生成3'突出端,然后进行spri纯化;(2)通过酶促切割纳入的含有可切割碱基的pcr引物部分生成3'突出端;(3)通过指定量的标准化探针退火并连接至3'突出端保护指定数量的文库分子;和(4)5'外切酶消化无探针保护的文库部分。方法2a方法2a的进一步细节见图8a-8d、9a-9b和10a-10b;图8a描述了使用单5'突出端的反应,其中互补探针序列是复杂序列s(包含任意2、3或4个碱基组成,未显示)或是低复杂性的均聚物序列,例如但不限于图8a中所示的(dt)12。在该实例中,只有一个pre-n引物与常规反向引物联用,该pre-n引物使标准化反应仅靶向ngs文库双链中的一条链。在第一标准化步骤中包括过量的ngs文库,其中除了dna连接酶还添加限定摩尔量的n-探针。相较于高复杂性的序列s,低复杂性均聚物序列退火更快。由于使用核糖u碱基连续串作为复制挡块,5'突出端优选为多聚(t)均聚物,而n-探针优选是与t和核糖u碱基都互补的多聚(a)均聚物;在复制挡块由核糖a碱基组成的其他实施方式中,5'突出端优选为多聚(a),n-探针优选为多聚(t)均聚物。此外,除了任选的3'末端块如c3碳间隔区,n-探针包含1种或多种核酸酶抗性修饰如硫代磷酸酯键连接,所述修饰可以在探针序列内连续地位于在其3'末端、内部或其5'末端附近,从而在探针退火和连接至文库末端后提供对于外切核酸酶iii消化的抗性。任选地,n-探针的长度比5'突出端长,从而在连接到ngs文库后产生单链3'突出端,为的是赋予对具有双链dna特异性活性的外切核酸酶iii消化的进一步抗性。限定摩尔量的n-探针退火连接后,进行第二标准化步骤,其中,添加外切核酸酶iii以消化过量的无探针保护的文库部分。由于其dsdna特异性3'至5'外切核酸酶活性,完全未保护的文库分子从3'末端开始消化,直至与相对链的消化相遇,产生无功能并且不能进行扩增或测序的两个单链部分文库片段。对于ngs文库单链保护的部分,未保护的链被完全消化,而被探针保护的链是核酸酶抗性的。因此,需要2倍摩尔过量的ssdnan-探针来保护相应指定摩尔量的dsdna文库,其中所得文库是单链的并且就衔接子序列而言定向(图8b)。例如,在图8b中,8nm的标准化探针将产生8nm的单链dnangs文库,相当于4nm的双链dnangs文库。或者,可以使用两个pre-n引物进行方法2a,在ngs文库的两个末端产生5'突出端,从而在标准化后回收两条链(图8c和8d)。在该实例中,从过量的文库到n-探针摩尔浓度,回收的所需量的dsdna文库也需要使用两倍摩尔浓度的ssdnan-探针。虽然回收了相应的dsdna文库量,但是经标准化的文库含有ssdna和dsdna文库分子,这取决于各双链体中是一条链还是两条链被保护,因为n-探针相对于输入ngs文库而言是有限的。例如,在图8c中,需要8nm单链探针和超过4nmpcr扩增的dsdna文库以产生4nm的dsdnangs文库量。对于方法2a,可以使用2种或4种引物进行使用pre-n引物的pcr扩增(分别为图9a和10,和图9b和图10b)。当仅靶向一个ngs文库链以在每个dsdna文库分子产生单5'突出端时,在2引物pcr中,一个pre-n引物与不具有5'尾序列的常规反向引物一起使用。虽然图9a-9b显示引物对中两种引物都包含纳入了标引序列的长5'尾并且当连有截短ngs衔接子的文库用作模板时形成全长衔接子(并且其中pre-n引物导入包含n-探针退火尾区的另一5'尾区),但是这两种引物也可以设计成扩增完整的、全长含标引的文库,其中引物仅退火结合用于文库扩增的末端衔接子序列,并且只有pre-n引物包含具有n-探针退火尾区的5'尾序列,而常规引物不具有5'尾(未显示)。4引物pcr扩增(图9b)限于连有截短ngs衔接子的文库作为模板,不是在pre-n和常规引物上包含长5'尾以形成全长ngs衔接子序列并同时导入用于n-探针退火的尾区,序列纳入是依次进行的,其中,将第一标引引物对用于形成全长ngs衔接子,两个引物都含有形成全长ngs衔接子的5'尾,然后第二个pre-n引物对将用于n-探针退火的尾区导入到第一引物对的扩增产物上,在这种情况下,pre-n引物与常规引物联用以靶向ngs文库的一条链。在4引物反应中,标引引物对可以较低浓度使用以促其消耗,而pre-n引物对则以较高浓度用于文库扩增以减少成品文库产物中出现第一引物对产物的可能性(这会消除用于n-探针退火的尾区)。虽然图9a-9b仅显示用于生成靶向单链的5'突出端的第一ngs衔接子取向,但是相对的,可用第二ngs衔接子生成5'突出端,然后将常规引物用于第一ngs衔接子(未显示)。如图10a-10b所示,当设计方法2a靶向ngs文库的两条dna链时,无论是使用2引物还是4引物,在两种情况下都存在两个pre-n引物,两者都导入具有相同组合的5'末端尾区,但这两个引物各自特异性地退火结合第一或第二ngs衔接子序列并且任选地包含衔接子序列的末端部分,由此在文库的两个末端生成对称的5'突出端,而不是使用pre-n和常规引物对。在图9b和10b中,pcr反应以标引引物开始,并且当标引引物耗尽时由标准化引物继续反应(例如,如图9b和10b所示,在10nm的ngs文库浓度)。方法2b方法2b的进一步细节见图11,其中由t4dna聚合酶生成5'突出端;该图显示用具有5'缓冲区的常规反向引物与一个pre-n引物对文库一条链进行外切核酸酶保护,但是如果使用两个pre-n引物,对文库两条链进行外切核酸酶保护也是可能的(未显示)。在图11中,在pcr扩增步骤中,仅一个包含5'末端尾区和3'侧缓冲区的pre-n引物与包含5'末端缓冲区的反向引物一起使用,反向引物5'末端缓冲区与第一缓冲区在碱基组成上相同。缓冲区的碱基组成限于尾区所没有的核苷酸。在生成相对于待用n-探针摩尔量过量的文库后进行纯化步骤,此后进行第一标准化步骤:添加限定摩尔量的与尾区互补的n-探针以及t4dna聚合酶,与缓冲区互补但是不与尾区互补的核苷酸混合物和dna连接酶。优选使用低复杂性n-探针序列,如均聚物,二核苷酸或三核苷酸序列,因为低复杂性探针与互补突出端退火结合比高复杂性序列快。此外,n-探针包含1种或多种核酸酶抗性修饰,如硫代磷酸酯键,所述修饰可以在探针序列内连续地位于其3'末端、内部或其5'末端附近,从而在n-探针退火和连接至一个或多个文库末端后提供抵抗外切核酸酶iii消化的抗性。它还包含保护基团,如3'磷酸或二级结构,如3'自身互补的发夹和复制挡块的组合,或赋予n-探针抵抗t4dna聚合酶3'外切核酸酶活性的抗性的任何其他修饰。在这第一个酶法标准化反应中,t4dna聚合酶在先前的dsdna尾区生成5'突出端以使n-探针能退火结合扩增文库,其中,在适当的反应条件下,t4dna聚合酶不可逆地从互补尾区去除3'碱基,反应中没有所述互补尾区的dntp,但是由于存在与缓冲区互补的dntp,t4dna聚合酶可逆地去除并置换3'侧缓冲区的碱基。扩增文库的相对末端由于存在与第一缓冲区具有相同核苷酸组成的第二缓冲区因此保持双链状,由于存在相应的核苷酸混合物,其中的碱基被t4dna聚合酶可逆地去除并替换。这由此限定了第一末端在缓冲区的5'突出端,在相对的末端,第二缓冲区防止形成突出端。限定量n-探针退火并连接后,添加外切核酸酶iii消化过量的无探针保护文库部分来进行第二个酶法标准化步骤。由于其dsdna特异性3'至5'外切核酸酶活性,完全未保护的文库分子从3'末端被消化,直至与相对链的消化相遇,由此产生无功能并且不能进行扩增或测序的两个单链的部分文库片段。对于ngs文库单链保护的部分,未保护的链被完全消化,而被探针保护的链是核酸酶抗性的。因此,需要2倍摩尔浓度过量的ssdnan-探针量来保护相应的dsdna文库量,其中所得文库是单链的并且就衔接子序列而言定向(图11)。在该方法的另一实施方式中,不添加外切核酸酶iii来消化非探针选定部分,而是可以添加三磷酸腺苷双磷酸酶二磷酸酶(apyrasediphosphataseenzyme)来水解反应中dntp混合物中的磷酸基团,然后这在不存在用于替换被切除碱基的任何功能性dntp的情况下,导致t4dna聚合酶不受控制的3'-5'外切核酸酶活性,因此,非探针选定文库部分被消化并变得不可测序,而n-探针选定部分则保持核酸酶抗性并且可测序。在其他方面中,可以使用各自都含有尾区和缓冲区的两个pre-n引物进行方法2b,在ngs文库的两个末端生成5'突出端,从而在n-探针连接和文库消化后回收ngs文库的两条链。在一实施方式中,均聚物尾区与二核苷酸缓冲区联用,但是任何低复杂性序列都可以用于尾区,只要缓冲区的碱基组成是尾区所没有的。可以使用线性n-探针,其在其3'末端、内部或其5'末端附近包含核酸酶抗性修饰,或者可以使用具有3'自身互补发夹和不可复制间隔区的任选的n-探针赋予对t4dna聚合酶3'外切核酸酶活性和外切核酸酶iii的进一步抗性。在两种情况中,探针赋予对3'外切核酸酶消化的抗性。在这两个酶法标准化步骤之后,从相对于n-探针摩尔浓度过量的文库输入中回收所需摩尔量的dsdna文库,但使用单链dna两倍摩尔浓度的n-探针。虽然回收了dsdna摩尔文库量,但是标准化的文库含有ssdna和dsdna文库分子,这取决于各双链体中是一条链还是两条链受到保护,因为n-探针是有限的。方法2c该方法见图12,其中用于n-探针连接的3'突出端在pcr后通过切割酶生成。虽然该附图仅显示使用一个可切割的pre-n引物在ngs文库一条链上生成单3'突出端,但是如果两个pcr引物都含有可切割的碱基,那么可以在文库两个末端生成3'突出端,由此在退火和连接核酸酶抗性n-探针后保护ngs文库两条链免于5'外切核酸酶消化。pre-n引物中包含的可切割碱基可以是rna、脱氧尿嘧啶或脱氧肌苷,其中纳入的引物序列可以分别被rna酶h,尿嘧啶dna糖基化酶和脱嘌呤内切核酸酶或内切核酸酶v从所得扩增子上切掉。在生成相对待使用的n-探针摩尔量过量的ngs文库后的纯化步骤之后,除了切割酶和dna连接酶之外,通过添加指定摩尔量的n-探针进行第一标准化反应。优选使用与3'突出端互补的低复杂性探针序列,如均聚物、二核苷酸或三核苷酸序列,因为低复杂性探针退火比高复杂性序列更快。此外,n-探针包含1种或多种核酸酶抗性修饰,如硫代磷酸酯键,所述修饰可以在探针序列内连续地位于其5'末端、内部或其3'末端附近从而在探针退火和连接至一个或多个文库末端后提供对于5'外切核酸酶消化的抗性。在该反应中,pre-n引物的长度和可切割碱基的位置和数量决定酶促切割生成的3'突出端的长度。因此,切割酶生成3'突出端以使探针能够使有限的、指定量的探针与过量的3'突出端退火并连接,这赋予选定文库部分5'外切核酸酶抗性。在第二个标准化步骤中,然后添加λ或t7外切核酸酶来消化无探针保护的文库部分。由于其dsdna特异性5'至3'外切核酸酶活性,完全未保护的文库分子从5'末端被消化,直至与相对链的消化相遇,由此产生无功能并且不能进行扩增或测序的两个单链的部分文库片段。对于ngs文库单链保护的部分,未保护的链被完全消化,而探针保护的链是核酸酶抗性的。因此,需要2倍过量的ssdnan-探针来保护相应的所需dsdna文库量,其中所得文库是单链的并且就衔接子序列而言定向。图41a-41e还描述了用于受控保护免于外切核酸酶消化的不同示例性实施方式。在一些情形中,不需要突出端(参见例如图41a)。在一些情形中,连接可以是例如但不限于钝端(图41a)、ta连接(图41b)或粘性末端连接。在图41a-41e中所示描述的所有方法中,携带核酸酶抗性碱基的限定定量的n-探针可以连接到3'或5'或两个dna末端,从而保护指定文库部分免于外切核酸酶降解。文库分子可以经由本文所述和本领域技术人员所知的方法产生。在通过pcr产生突出端的情形中,本文所述方法可用于生成这类突出端。在图41a中,以高保真dna聚合酶和常规引物扩增ngs文库产生钝端片段,然后可以将其钝端连接至指定量具有修饰的双链(或单链,未显示)探针,所述修饰提供对外切核酸酶消化的抗性,然后暴露于外切核酸酶以产生标准化的ngs文库。为了效率,可以非常高的文库和dna连接酶浓度进行反应,因为不存在低复杂性序列来加快探针退火和连接。在图41b中,以taq或没有3'校对活性的任何其它热稳定性dna聚合酶和常规引物扩增文库,产生末端具有a尾的片段,然后可以通过ta连接将其连接至具有t尾的标准化探针。对于图41a,为了效率,可以非常高的文库和dna连接酶浓度进行反应,因为不存在低复杂性序列来加快探针退火和连接。在图41c中,使用pre-npcr引物扩增文库以纳入5'尾和3'侧缓冲区。尾区是6-20个碱基并且包含均聚物、二-或三-核苷酸组成,后跟5-10个碱基的缓冲区,其包含5'尾区中所没有的核苷酸组成。当用t4dna聚合酶以及限于仅与缓冲区而非尾区碱基互补的核苷酸混合物孵育包含这类序列的ngs文库时,t4dna聚合酶的3'外切核酸酶校对活性将会不可逆地修整3'互补尾区直到其到达缓冲区,在此,其可以可逆地去除并替换核苷酸,因此产生由缓冲区限定的5'突出端。产生5'突出端并将n-探针与互补的5'突出端序列连接可以在去除或加热灭活t4dna聚合酶后依次进行,或当n-探针5'尾区的3'侧具有相同碱基组成的缓冲区时,合并成单个孵育反应。pre-npcr引物和n-探针互补尾序列是均聚物重复序列,如多聚a和多聚t,由此加快探针退火和连接,从而实现低探针浓度下的有效连接。在图41d中,文库使用具有5'尾和5'尾与衔接子序列之间的不可复制基团或不可复制间隔区的pre-npcr引物扩增,所述不可复制基团包含du碱基(针对古细菌(archael)dna聚合酶),所述不可复制间隔区含有3个或更多个核糖u或核糖a碱基的连续串,高保真dna聚合酶不能通过(核糖u)n或(核糖a)n模板延伸,其中n=3或更多。当核糖u和核糖a碱基分别侧接多聚(dt)和多聚(da)6-20碱基尾序列时,pcr产生的文库分子具有相应的均聚物5'突出端用于均聚物n-探针连接。重复序列如多聚a和多聚t加快探针退火和连接,从而实现低探针浓度下的有效连接。在图41e中,使用具有经修饰的碱基的pre-npcr引物扩增文库,所述经修饰的碱基包括可切割碱基或核酸酶抗性碱基。可切割碱基可以均匀地位于5'尾内或衔接子序列的5'部分内,并且包括但不限于du碱基、rna和脱氧肌苷。核酸酶抗性碱,如硫代磷酸化碱基可以位于5'尾的3'部分内,或者如果尾不存在(未显示)则位于衔接子序列内。以udg/脱碱基内切核酸酶混合物、rna酶h或内切核酸酶v(在可切割碱基的情况下)和5'外切核酸酶如t7外切核酸酶、λ外切核酸酶或外切核酸酶v(在核酸酶抗性碱基的情况下)孵育将产生3'突出端,其长度由pre-npcr引物内可切割/核酸酶抗性碱基或连接的位置控制。尾序列可以是复杂序列或均聚物低复杂性序列。方法3:通过受控修复进行酶法文库标准化图13-16中概述了3种受控修复方法,其中在pre-n引物pcr扩增之后,方法3a具有两个标准化步骤:切割/连接,然后是外切核酸酶步骤,而方法3b和c仅具有单个切割/连接步骤。与方法2c类似,方法3a和b利用含有可切割碱基的pre-npcr引物生成过量摩尔量的pcr后ngs文库上的3'突出端,而方法3c使用5'外切核酸酶和含有核酸酶抗性修饰的pre-n引物生成3'突出端。在方法3a中,使用一个pre-n引物和常规引物,产生的单3'突出端包含ngs衔接子的功能性部分,因此切割纳入的引物序列导致50%文库分子的部分文库失活,因为没有衔接子5'部分,切割后的文库链就不能在illumina测序平台形成簇或在离子仪器(ioninstrument)上支持乳液pcr,但第二ngs链保持完整和功能性。如果使用两个pre-n引物而不是一个(方法3b),那么文库的两条链或100%的文库分子都会被切割反应灭活。在这两种方法中,标准化引物都包含rna、脱氧尿嘧啶或脱氧肌苷碱基,与rna酶h、udg和脱嘌呤内切核酸酶或内切核酸v切割联用。3'突出端的长度由引物内可切割碱基的位置以及整体引物长度控制。在其他实施方式中(方法3c),在pcr后通过与5'外切核酸酶如t7或λ外切核酸酶孵育在一个或两个文库末端生成3'突出端,所述孵育将消化扩增文库分子的5'末端直到在内部遇到核酸酶抗性连接如硫代磷酸酯键或其他修饰。3'突出端的长度由pre-npcr引物内可消化碱基或核酸酶抗性碱基或连接的位置以及整体引物长度控制。酶法标准化步骤还包含有限摩尔量的n-探针和连接酶。任选地,还包括如taqdna聚合酶或侧翼内切核酸酶之类的酶来置换和切割引物消化反应不完全而留下的任何残留的可切割碱基,否则这些残留的可切割碱基将干扰n-探针完全退火和连接。在该步骤中,在酶促去除任一个(方法3a)或两个(方法3b和c)5'末端以使一条或两条文库链失活后,将过量摩尔量的文库的选定部分连接至有限摩尔量的n-探针,所述n-探针对应通过切割去除的ngs衔接子序列,由此恢复功能性ngs衔接子及其接受测序的能力。这完成了方法4b和c的酶法标准化过程,因为恢复了功能的选定文库部分是对应n-探针有限摩尔量的摩尔量。方法3a还需要一标准化步骤,其中使用单链特异性3'至5'外切核酸酶如外切核酸酶i消化文库链上保持完整但其互补链不与n-探针连接的3'突出端,由此灭活除n-探针连接文库链之外的所有文库链,这将ngs文库的摩尔浓度标准化以对应n-探针摩尔量。具体地,在图13中,有4个步骤描述了这3种方法:(1)进行pcr扩增以生成在一个(方法3a)或两个(方法3b或3c)衔接子末端具有可切割碱基(方法3a或3b)或核酸酶抗性碱基(方法3c)的ngs库,然后进行spri纯化;(2)通过消化ngs衔接子之一的5'部分使部分文库失活(方法3a)或通过酶促消化两个ngs衔接子的5'部分(方法3b)使全部文库失活,或采用5'外切核酸酶消化(方法3c),生成3'突出端;(3)通过退火和连接指定量的标准化探针至3'端使指定数量的文库分子恢复;和(4)通过使用单链特异性外切核酸酶如外切核酸酶i消化3'突出端使未修复的文库部分完全失活(仅限方法3a)。方法4:通过受控合成进行酶法文库标准化图17中概述了用于酶法文库标准化的两种受控合成方法,其需要构建和扩增这样的ngs文库,所述ngs文库在一端或两端具有截短的或不完整的、非功能性衔接子序列,然后连接限定的n-探针数量,这完成文库选定摩尔部分的功能性文库构建。在方法4a中,在pcr期间使用pre-n引物中的不可复制部分生成5'突出端,或在方法4b中,如前所述,在pcr后通过t4dna聚合酶生成5'突出端(pre-npcr引物缓冲区不包括任何尾中的碱基组成,以及此后pcr后3'-5'外切核酸酶反应中互补尾区中没有而互补缓冲区中有的限定dntp组成)。在其他方面中,5'突出端可以通过本文其他方法生成。或者,可以在通过方法2c中就n-探针退火和连接所述的任何方法(未显示)生成3'突出端后进行受控合成标准化。在形成5'或3'突出端后,在一实施方式中(方法4a),使用单链n-探针仅将文库功能性赋予其连接的链。在另一实施方式(方法4b)中,使用部分双链的n-探针将文库功能性赋予其连接的两条链。形成5'或3'突出端并非n-探针连接的必要条件,但是突出端显著促进低浓度时探针和文库连接。在一实施方式中,带单t-碱基3'突出端的双链标准化探针的连接需要在pcr期间通过taqdna聚合酶产生的具有单a-碱基3'突出端的文库(图42b)。在另一实施方式中,双链标准化探针具有钝端,并且连接使用高保真dna聚合酶扩增的文库(图42a)。pcr期间产生的高文库浓度可以促进限定量的钝端或单t-碱基3'突出标准化探针与截短衔接子末端的连接。阻止探针连接至ngs文库相对端的衔接子可以通过缺少5'磷酸基团或缺少相容钝端或单a碱基突出端或同时缺少5'磷酸和相容末端来控制。具体地,在图17中,显示的两个步骤包括:(1)使用含有不可复制碱基或碱基组合的pcr引物进行pcr扩增在截短衔接子末端生成具有5'突出端的非功能性文库分子(方法4),或在两端用特殊的碱基组成从而能够通过t4dna聚合酶获得5'突出端生成(方法4),然后进行spri纯化;和(2)通过指定量的标准化合成探针退火和连接5'突出端合成指定数量的功能性文库分子(方法4)或者首先,在限定核苷酸组成存在下由t4dna聚合酶在截短衔接子末端产生5'突出端,然后指定量的双链标准化合成探针退火连接5'突出端(方法4)。图18-42i显示了当基于受控合成的标准化靶向一个或两个文库链时的细节。图18、20和42d描述了当以单个pre-n引物组合常规反向引物靶向一个文库链时的可选方案,所述单个pre-n引物保留截短的第一ngs衔接子但引入低复杂性(包括但不限于均聚物)探针退火位点作为5'突出端,所述常规反向引物产生钝端并且可以导入样品标引,如illumina测序所需的。pcr后,在将pcr产物与n-探针和dna连接酶孵育之前,进行纯化步骤。在这种情况中,n探针包含与低复杂性序列互补的序列,并在连接时导入截短ngs衔接子缺失的部分以形成功能性ngs文库。所得文库包含具有内部低复杂性插入的全功能性衔接子。在另一实施方式中(图42e-42h),5'突出端探针退火区域对应功能性衔接子序列,而不是导入低复杂性序列以促进快速退火。在这种情况中,可以通过用核糖核苷酸替换衔接子序列内的一些核苷酸来产生5'突出端。在一个这类实施方式中,pre-n-引物内的三个连续胸腺嘧啶碱基被三个连续的核糖尿苷碱基替换(图42e和图43b-43)。在这种情况中,在n-探针连接后,生成不包含低复杂性序列的功能性ngs文库。在另一实施方式中,可以通过添加彼此互补的低复杂性序列例如多聚a和多聚t分别至pre-n引物和n-探针的5'和3'端来加快包含衔接子序列的单链n-探针的连接(图42g和43c)。在这种情况中,在n-探针连接后,产生的功能性ngs文库在衔接子的末端而非衔接子内部包含低复杂性序列。在将有限摩尔量的单链或双链n-探针与所得5'突出端连接后,产生对应n-探针摩尔浓度的单链功能性文库。应当理解的是,需要两倍高的ssdna探针浓度以生成指定摩尔浓度的dsdna文库当量。图19和图21a描述了当以两个pre-n引物靶向两条文库链时的可选方案,其中一个pre-n引物保留截短的第一ngs衔接子但导入促进t4dna聚合酶介导的5'突出产生的其他序列,而第二pre-n引物在第二ngs衔接子末端导入标引序列和缓冲区赋予对t4dna聚合酶3'外切核酸酶活性的抗性以维持钝端。用pre-n-引物进行pcr扩增后,然后纯化去除未使用的引物并进行缓冲液交换,用t4dna聚合酶和限定的脱氧核苷酸组合孵育纯化的pcr产物,所述限定的脱氧核苷酸组合与缓冲序列互补并且与尾部序列不互补,从而生成在截短的第一衔接子末端具有5'突出端并在完整的第二衔接子末端具有钝端的产物。在t4dna聚合酶热灭活后,将有限摩尔量的n-探针连接至5'突出端,所述n-探针包含与低复杂性尾序列互补的序列以及赋予功能性所需的ngs衔接子的缺失部分。在另一实施方式中,n-探针在其3'末端还包含3'外切核酸酶抗性的缓冲区,从而能够同时进行t4dna聚合酶产生5'突出端和连接。图20a-20b显示了当使用单个pre-n引物(方法4a)时,任一衔接子末端都可以用于导入用于n-探针退火和连接的5'突出端;另外,无论是针对第一或第二衔接子端的标准化,都可以包括单标引或双重标引。在图21a中,pcr扩增的ngs文库包含带标引的全长p7衔接子和截短的p5衔接子,其中p5具有末端探针退火和缓冲序列,而p7具有缓冲序列(缓冲序列配合相应的dntp组成阻断t4dna聚合酶外切核酸酶消化)。在图21b中,全合成ngs文库具有p7标引以及任选的p5双重标引,其中p5衔接子包含内部标准化探针序列。在另一实施方式中,通过受控合成进行标准化不需要产生截短的衔接子文库。这是因为由于核糖核苷酸与测序工作流程的不相容性,将核糖核苷酸碱基插入衔接子序列可以使文库无功能性。因此,如果全长衔接子包含内部核糖核苷酸,那么它可以是无功能的。如图42h所示,具有核糖碱基的pre-n引物可具有全长衔接子序列,但是至少对于illumina测序平台而言,pcr产物仍然可能是无功能的,因为pcr产物的一条链(下方)被截短并且是无功能的,而另一条(上方)链不能用illumina流动池上簇形成所用高保真dna聚合酶完全复制,因此其也是无功能的。随后将n-探针连接到pcr产物的底链产生功能性文库,并且该文库的量由标准化探针的量控制。在一个方面,用于基于pcr或酶法ngs文库标准化的试剂盒包括进行本文所述方法或方法的组合及其实施方式所需的组件。在另一方面中,用于酶法ngs文库标准化的试剂盒包括方法2a所需的组件,包括pre-n引物,聚合酶,n-探针,连接酶和核酸酶。在另一实施方式中,用于基于pcr的文库标准化的试剂盒包括n-pcr引物对和聚合酶。用于受控合成的其他方法可见图42a-42h,其中包括这样的实施方式:用多种方法中任选的方法产生在一个末端具有截短衔接子的截短ngs库,而探针提供衔接子的缺失部分以恢复ngs文库进行ngs所需的功能。在图42a,用高保真dna聚合酶和常规引物扩增文库以产生钝端片段,然后连接到钝端n-探针。为了效率,这类反应应当以非常高的文库和dna连接酶浓度进行,以便能够在不存在低复杂性序列的情况下以低探针浓度连接。通过将庞大间隔区置于相应pcr引物的5'末端,无缺陷末端可以避免这类连接。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。在图42b中,以taq或缺少3'校对活性的任何其它热稳定性dna聚合酶和常规引物扩增文库,产生在末端具有a尾的片段,然后连接具有t尾的n-探针。如在情况a中,这类反应应当以非常高的文库和dna连接酶浓度进行,以便能够在不存在低复杂性序列的情况下以低探针浓度连接。通过将庞大间隔区置于相应pcr引物的5'末端,无缺陷末端可以避免这类连接。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。在图42c中,使用pre-npcr引物扩增文库在ngs文库截短衔接子末端纳入5'尾和3'侧缓冲区。尾区是6-20个碱基并且包含均聚物、二核苷酸组成,后跟5-10个碱基的缓冲区,缓冲区的核苷酸组成是5'尾区中所没有的。当用t4dna聚合酶以及限制为仅与缓冲区而非尾区碱基互补的核苷酸混合物孵育包含这类序列的ngs文库时,t4dna聚合酶的3'外切核酸酶校对活性将会不可逆地修整3'互补尾区直到其达到缓冲区,在此则可逆地去除并替换核苷酸,因此产生由缓冲区限定的5'突出端。产生5'突出端并将n-探针与互补的5'突出端序列连接可以在去除或加热灭活t4dna聚合酶后依次进行,或当n-探针含有5'尾的3'侧相同碱基组成缓冲区时,合并成单个孵育反应。在最佳的情况下,pre-npcr引物和n-探针互补的尾序列是均聚核苷酸(homo-nucleotide)重复序列,如多聚a和多聚t,由此加快低探针浓度时的探针退火和连接。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。在图42d中,文库使用具有5'尾和位于5'尾和衔接子序列之间的不可复制基团或不可复制间隔区的pre-npcr引物扩增,所述不可复制基团包含du碱基(针对古细菌(archael)dna聚合酶),所述不可复制间隔区含有3个或更多个核糖u或核糖a碱基的连续串,高保真dna聚合酶不能通过(核糖u)n或(核糖a)n模板延伸,其中n=3或更多。当核糖u和核糖a碱基分别侧接多聚(dt)和多聚(da)6-20碱基尾序列时,pcr产生的文库分子具有相应的均聚物5'突出端,用于与具有均聚物5'尾的n-探针连接。重复序列如多聚a和多聚t加快探针退火和连接,从而能够以低探针浓度有效连接。扩增所得文库最初是具有缺陷型衔接子末端的经截文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。在图42e中,使用pre-npcr引物扩增文库,其中用3个或更多个核糖碱基的连续串替换原ngs衔接子序列内的脱氧核苷酸。n-探针是双链的。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。当使用不具有3'校对活性的聚合酶时,没有向截短的衔接子序列添加其他序列,该方法能够生成具有5'突出端的截短文库。在图42f中,使用pre-npcr引物扩增文库,其中用3个或更多个核糖碱基的连续串替换原ngs衔接子序列内的脱氧核苷酸。n-探针是单链的。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。在图42g中,使用pre-npcr引物扩增文库,其中用3个或更多个核糖碱基的连续串替换原ngs衔接子序列内的脱氧核苷酸且5'末端具有均聚-,二-或三-核苷酸重复序列。n-探针是单链的,并在5'末端具有互补的重复序列,从而在低探针浓度加速探针退火和连接。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。n-探针在其3'末端可以具有10-20个多聚a或多聚t碱基。因此,在一些实施方式中,截短ngs文库可以包含突出端,一些情况中是5'突出端,这些突出端还包含本文所述的低复杂性序列来加快退火以利文库扩增,同时允许通过探针当所述探针与截短的ngs文库连接时重新纳入缺失的衔接子部分(存在于环中)。这可以恢复截短ngs文库中与探针连接的那些文库分子的功能,但因为低复杂性序列和探针上互补序列之间的结合,所以速率更快且更有效。在图42h中,使用具有经修饰的碱基的pre-npcr引物扩增文库,所述经修饰的碱基包括可切割碱基或核酸酶抗性碱基。可切割碱基均匀地位于衔接子序列的5'部分内,并且包括但不限于du碱基、rna和脱氧肌苷。核酸酶抗性碱基(如硫代磷酸化碱基)可以位于5'尾的3'部分内,或者如果不存在尾则位于衔接子序列内。以udg/脱碱基内切核酸酶混合物、rna酶h或内切核酸酶v(在可切割碱基的情况下)和5'外切核酸酶如t7外切核酸酶、λ外切核酸酶或外切核酸酶v(在核酸酶抗性碱基的情况下)孵育将产生3'突出端,其长度由pre-npcr引物内可切割/核酸酶抗性碱基或连接的位置控制。产生5'突出端并将n-探针与互补的5'突出端序列连接可以在去除或加热灭活切割酶后依次进行,或合并成单个孵育反应。如果是外切核酸酶消化,n-探针在3'突出端远端的末端部分内含有具有核酸酶抗性碱基的保护区。扩增所得文库最初是具有缺陷型衔接子末端的截短文库,所述缺陷型衔接子末端连接至包含衔接子缺失部分的探针而产生功能正常化的ngs文库。n-pcr:基于pcr的ngs文库标准化方法通过pcr(n-pcr)标准化ngs文库浓度依赖于以下假设:所有pcr引物可在扩增期间被利用并转化为ngs文库,从而产生摩尔量等于n-pcr引物摩尔量的ngs文库。为了确保有效扩增ngs文库并保持其复杂性,常规pcr反应中的引物浓度通常在200nm至>1,000nm之间变化。使用该引物浓度,假设pcr引物被完全利用,在50μl反应体积中进行的pcr扩增反应应当产生30-50pmol的ngs文库。生产这样体量的文库将需要更多的dna聚合酶,这是不切实际的,但即便如此,反应仍会在后期pcr循环中受到大量dna抑制的影响。这还需要更多的pcr循环以将所有引物转化到文库,显著增加pcr重复和dna过度受热有损害dna并导入热诱导突变的风险。该问题或许可以通过降低引物浓度来解决,但当引物浓度降低时,如果使用标准退火和延长时间,那么引物引发效率和文库复杂性会较低;或者,如果增加退火和延伸时间以补偿引物浓度降低,pcr反应可能将持续数小时。基于pcr的文库标准化可以通过使用本文所述新型引物设计来实现,该新型引物设计在低引物浓度也能使引物高效退火,因此能够使用常规的退火时间。这些经修饰的n-pcr引物可以在低浓度和用常规退火时间进行而不会降低引物引发效率或ngs文库复杂性。加速退火过程的示意图示于图22中。退火时间加快的经修饰的n-pcr引物具有两个结构域:3'结构域,其退火结合ngs衔接子序列,用于文库扩增,以及加速退火并减少退火时间的5'结构域,这是相比常规文库扩增引物多出的特征。5'结构域包含具有低序列复杂性的dna序列,如单-、二-、三-、四-核苷酸重复。当dna底物具有与引物5'结构域重复序列互补的低复杂性重复序列(以及与引物3'部分互补的序列)时,引物通过其5'重复结构域与模板中互补的对应重复序列快速退火结合,这一锚定作用使得引物的3'结构域退火结合更高复杂性的序列。5'结构域锚定形成局部的高引物浓度并导致引物3'结构域的快速退火和延伸。存在有许多简单重复序列可以用于高退火速率的n-pcr引物5'结构域,但并不是所有的都可用。为了避免n-pcr引物之间的交叉相互反应,5'结构域序列应选自两种非互补的重复序列,例如,就均聚物序列而言的多聚a和多聚c,或多聚t和多聚g。就二核苷酸重复而言,引物对的5'结构域序列选自两个非互补序列,如多聚gt和多聚ga,多聚gt和多聚ct,多聚ag和多聚ca,或多聚ct和多聚ca。选择互补尾序列如多聚a和多聚t,或多聚gt和多聚ca是不可取的,因为这将导致引物的5'结构域彼此退火结合并降低加速效应。5'结构域序列还可以选自三核苷酸、四核苷酸、五核苷酸和六核苷酸重复组合,同理需避免5'结构域彼此之间互补。在5'端具有相同重复序列(如多聚a,多聚gt,多聚acg等)的n-pcr引物将导致扩增子链折叠成自我互补的茎-环结构,还会导致两pcr引物竞争相同的重复结合位点。因此,n-pcr引物的5'结构域还可以包含非重复部分,如果这部分是其他功能所必需的。图23a显示了在n-pcr引物的5'末端使用多聚(ga)和多聚(gt)尾的示例性方法,得到了限定引物浓度的受控扩增。图23b显示了使用多聚(a)和多聚(t)尾用于相同目的示例性方法。图24a显示了引物和文库摩尔浓度随时间变化的假设图。图24b显示了三种不同文库输入浓度的相同类型的图。图25提供了具有均聚物尾、二核苷酸尾和三核苷酸尾的n-pcr引物的实例。除了n-pcr引物上的低复杂性5'结构域外,相应的dna模板应当具有与引物5'末端重复序列互补的重复序列,以允许n-pcr引物与模板快速退火。可以在文库合成期间通过连接含有重复序列的ngs衔接子将这些序列添加到ngs文库末端。优选此法因为它允许文库扩增时无需在前两个循环中进行长退火时间来通过pcr纳入低复杂性尾序列。或者,当文库在衔接子末端不具有低复杂性序列时,可以在文库扩增期间使用含有重复序列的引物导入重复序列。后一种情况中,至少应延长前两个pcr循环的退火时间以确保在dna模板中还没有互补重复序列时引物能完全退火(和延伸)。2个pcr循环后,将生成在两端含有重复序列的文库扩增子,即可使用较短的常规退火时间继续pcr循环。图26显示了提出的n-pcr引物加速退火机制。图27和28显示了使用n-pcr引物进行扩增和标准化的示例性方法。图29a显示了具有均聚物尾序列(最低复杂性)的n-pcr引物应当有可能提供退火的最大加速,但是使用这种尾序列可能出现某些问题:具有相同均聚物尾的正向和反向引物产生具有茎环结构的扩增子,其中彼此互补的5'和3'尾形成茎区并且变得不易于发生引物延伸。即使它们能够与扩增子结合,但是这样的二级结构导致反向引物的均聚物尾与正向引物竞争结合,反之亦然。图29b显示了具有互补均聚物尾的正向和反向n-pcr引物产生不形成二级结构的扩增子,但是引物本身可以通过它们的尾序列相互作用,这使得它们不能结合。图29c显示了具有非互补均聚物尾(如多聚(t)和多聚(g),多聚(t)和多聚(c),多聚(a)和多聚(g)或多聚(a)和多聚(c))的n-pcr引物产生扩增子,其中5'端或3'端具有多聚g序列,能够形成对引物和/或复制具有抗性的沃森-克里克二级结构(如g-四联体)。图29d显示了n-pcr引物的二核苷酸重复尾序列,这是优选并且可以使用的。然而,由于它们的互补性不应使用(ta)n和(gc)n。在一些实施方式中,n-pcr引物可以用于其它应用,例如,在诊断中用于加快病毒和细菌dna和rna模板的pcr扩增。当扩增非常低拷贝的病毒核酸需要30个pcr循环、约45分钟内(假设每个循环1.5分钟),使用本文所述引物的相同过程可以快近6.5倍,仅7分钟内即可完成。通过测量用至少一个5'端含荧光团的n-pcr引物扩增所得文库的荧光强度,可以容易地确定标准化反应的成功以及n-pcr后ngs文库的摩尔浓度。在添加过量摩尔量的猝灭寡核苷酸之前和之后进行这样的测量,所述猝灭寡核苷酸与含荧光团的n-pcr引物或其5'部分互补并且在3'端含有猝灭发色团(如果两个引物都具有荧光团则需两个猝灭寡核苷酸)。该方法优于使用荧光嵌入染料的常规浓度测量的一个优点是该方法在计算摩尔浓度时与插入大小无关,因此对于插入大小范围较宽或未知的底物,仍然可以精确测量摩尔量。另一个优点是这种定量方法不需要在测量之前去除pcr引物,因为它可以区分已纳入的引物浓度与游离的引物浓度。在添加猝灭寡核苷酸之前进行第一荧光测量,确定由所有引物产生的总荧光信号f1,这包括已经pcr纳入文库的引物和仍留存于溶液中的引物。在添加猝灭寡核苷酸之后进行第二荧光测量,以确定源自纳入文库的引物的荧光信号f2。基于这两个测量值,文库分量比例和未利用的引物分量比例可以分别确定为:f2/f1和(f1-f2)/f1。扩增的ngs文库[文库]的相应摩尔浓度和未纳入的引物[引物]的摩尔浓度可以分别计算为:[文库]=f2/f1x[p0]和[引物(p)]=(f1-fl)/f2x[p0],其中[p0]是pcr反应开始时的引物摩尔浓度。图30显示了应用这类方法和计算以确定ngs文库摩尔浓度的示例性方法。上述公式基于两个假设:a)荧光团的量子产率在未纳入和纳入的引物中是相同的,和b)猝灭寡核苷酸与溶液中剩余引物退火结合彻底抑制它们荧光。通过在不存在和存在互补寡核苷酸的情况下实验测量pcr引物荧光强度测试并确认了第一个假设。至于第二个假设,众所周知,猝灭剂发色团对荧光团荧光的抑制可以是强的但未必彻底,对于真正的荧光团-猝灭剂对,在猝灭剂寡核苷酸退火后荧光强度降低约30-100倍。通过选择更好的荧光团-猝灭剂对和通过在3'末端使用具有多个猝灭剂基团的猝灭寡核苷酸,潜在地,这种降低能增加2-3倍。然而,既便当前的降低因数,无需除去未纳入的引物,也已能够非常准确地测评标准化反应的成功并且精确测量ngs文库浓度(参见实施例11)。实施例表1.引物延伸反应寡核苷酸表2.扩增寡核苷酸表3.结合动力学寡核苷酸表4.酶法文库标准化寡核苷酸序列id序列14tttttttttttt-rurururu-aatgatacggcgaccaccgaga15tttttttttttt-rurururu-caagcagaagacggcatacgagat16/5phos/a*a*a*a*aaaaaaaaaaaa表5.再结合动力学寡核苷酸表6.基于pcr的标准化pcr引物表7.标记的标准化pcr引物和淬灭剂其中,ru–核糖u,du–脱氧核糖u,ra–核糖a,rc–核糖c,rg–核糖g,*-硫代磷酸酯键,/3spc3/-3'端c3间隔区,/5iabkfq/-5'端iowafq淬灭剂,/36-fam/-3'端荧光素,并且其中/3iabkfq/-3'端iowafq淬灭剂,/56-fam/-5'端荧光素表8.通过合成法进行文库标准化的寡核苷酸表9.用于pacbio扩增子文库合成的寡核苷酸其中是/5phos/-5’磷酸基团,ru–核糖尿苷表10:ngs衔接子序列其中,xxxxxx是标引序列。表11实施例17的寡核苷酸序列id序列48ttttttttttttggcggcaattgcgatcgatgcactgtggcggcggc49gccgccgccacagtgcatcgatcgcaattgccgccaaaaaaaaaaaa50ttttttttttttggcgaattgcgatcgatgcactgtggcggcggc51gccgccgccacagtgcatcgatcgcaattcgccaaaaaaaaaaaa52ttttttttttttggaattgcgatcgatgcactgtggcggcggc53gccgccgccacagtgcatcgatcgcaattccaaaaaaaaaaaa54ttttttttttttgaattgcgatcgatgcactgtggcggcggc55gccgccgccacagtgcatcgatcgcaattcaaaaaaaaaaaa实施例1.在含有核糖核苷酸重复阻断子的dna模板上的引物延伸反应的示意图引物延伸反应由含有内部rna复制阻子并且3'端受保护的模板dna寡核苷酸组成,以阻止引物5'端的模板延伸。反应的另一组分是具有5'非互补末端的延伸引物(图31a-31b)。引物延伸反应后可以形成三种类型的产物。如果聚合酶的持续合成能力被完全抑制,那么不会观察到延伸,未延伸的引物将迁移到25b大小处(产物1)。如果引物延伸反应停在复制阻断子开始之处或其内部,那么可以观察到迁移在延伸引物上方但在模板下方的一种或多种延伸产物(产物2)。如果聚合酶能够绕过复制阻断子,将形成迁移在模板上方的产物(图31b)。实施例2.纳入dna模板的核糖尿苷串阻断校对dna聚合酶q5进行的引物延伸材料10μmru模板寡核苷酸(寡核苷酸1)10μmr(u)2模板寡核苷酸(寡核苷酸2)10μmr(u)3模板寡核苷酸(寡核苷酸3)10μmr(u)4模板寡核苷酸(寡核苷酸4)10μmr(u)6模板寡核苷酸(寡核苷酸5)10μmdu模板寡核苷酸(寡核苷酸6)10μm延伸引物(寡核苷酸10)热启动高保真2x主混合物(mastermix)(neb,目录号m0494s)低te缓冲液(天惠华公司(teknova)目录号to227)25bp梯标dna大小标志物(英杰公司(invitrogen),目录号10488-022)15%tbe-尿素凝胶(英杰公司,目录号ec68852box)sybr金染剂(英杰公司,目录号#s11494)方法在30μl反应体积中进行延伸反应,其中包含15μl2x热启动高保真2x主混合物,2μl的模板寡核苷酸,1μl的延伸引物和12μl的低te缓冲液。98℃加热反应45秒以活化酶,然后在60℃延伸3分钟。将样品在甲酰胺上样缓冲液中煮沸,并以200伏特在15%tbe-尿素凝胶上分离。凝胶用sybr金染剂染色,在dark读取灯箱(克莱尔化学研究公司(clarechemicalresearch))上观察并采用数码相机拍照。结果图32显示了通过热启动高保真酶在含有不同数量的核糖尿苷或脱氧尿苷作为复制阻断子的模板上延伸反应产物的电泳分析。泳道2显示了延伸引物的迁移,泳道3-5显示了ru、r(u)3和r(u)6模板的迁移。泳道6显示了在含有单个核糖尿苷的模板上引物的完全延伸,泳道7显示了在含有两个核糖尿苷的模板上的部分和完全延伸产物,泳道8-10显示在含有两个以上(3至6个)核糖尿苷的模板的仅部分延伸的产物。泳道11显示在含有单个脱氧尿苷的模板上通过q5酶完全阻断引物延伸。从泳道6可以看出,q5聚合酶可以完全绕过纳入模板中的单个核糖核酸,产生一个完整的延伸产物,其迁移在模板上方并由两个星号表示。当将多于一个的核糖尿苷纳入dna模板中时,观察到对q5酶所致引物延伸的显著影响。具体地,当将两个核糖核苷酸纳入延伸模板时,可以观察到非常少的完全延伸(泳道7)。当将多于两个核糖尿苷纳入模板寡核苷酸时,q5聚合酶不能绕过复制阻断子,仅产生部分延伸的产物,在凝胶上通过三个星号指示(泳道8-10)。如在泳道11中所示,纳入模板的脱氧尿苷能够完全阻断q5聚合酶的持续合成能力。结论纳入dna模板的核糖尿苷可以抑制校对聚合酶q5进行的引物延伸。为了引物延伸的有效阻断,复制挡块应当含有三个或更多个核糖尿苷。实施例3.在含有包括四种不同核糖核苷酸的复制阻断子的dna模板上使用不同热稳定性dna聚合酶进行引物延伸反应材料10μmr(u)3模板寡核苷酸(寡核苷酸3)10μmr(a)3模板寡核苷酸(寡核苷酸7)10μmr(c)3模板寡核苷酸(寡核苷酸8)10μmr(g)3模板寡核苷酸(寡核苷酸9)10μm延伸引物(寡核苷酸10)taq2x主混合物(neb,目录号m0270l)2xq5du绕路聚合酶(neb,目录号不可获得)kapahifi热启动readymix(kapa生物系统公司,目录号kk2601)gxldna聚合酶(克隆泰克公司(clontech),cat#r050a)热启动高保真2x主混合物(neb,目录号m0494s)低te缓冲液(天惠华公司目录号to227)25bp梯标dna大小标志物(英杰公司,目录号10488-022)15%tbe-尿素凝胶(英杰公司,目录号ec68852box)sybr金染剂(英杰公司,目录号#s11494)方法含2xtaq,2xq5du绕路聚合酶或热启动高保真2x主混合物的引物延伸反应在30μl反应体积中进行,所述反应体积含有15μl的主混合物,2μl的模板寡核苷酸,1μl的延伸引物和12μl的低te缓冲液。gxldna聚合酶进行的引物延伸在30μl反应体积中进行,所述反应体积包含6μl的5xprimestargxl缓冲液,2.5μl的dntp混合物(各2.5mm)2μl的模板寡核苷酸,1μl的延伸引物,1μl的primestargxldna聚合酶和17.5μl的低te缓冲液。95℃加热2xtaq聚合酶反应3分钟以活化酶,然后在60℃延伸3分钟。98℃加热2xq5du绕路聚合酶、热启动高保真2x主混合物或gxldna聚合酶反应45秒以活化酶,然后在60℃延伸3分钟。将样品在甲酰胺上样缓冲液中煮沸,并将20μl各反应在15%tbe-尿素凝胶上以200伏特分离。凝胶用sybr金染剂染色,在dark读取灯箱(克莱尔化学研究公司)上观察并采用数码相机拍照。结果图33a显示了taq酶和q5du绕路酶在含有不同核糖核苷酸作为复制阻断子的模板上延伸反应产物的电泳分析。泳道2和泳道7显示了延伸引物的迁移,泳道3-6显示了taq聚合酶在含有r(u)3、r(a)3、r(c)3和r(g)3串的模板上引物延伸反应的产物。如泳道3至6所示,taq聚合酶可以部分绕过由三核糖尿苷,三核糖腺苷或三核糖胞嘧啶组成的复制阻断子。引物延伸产生独特条带,对应复制停在复制阻断子开始之处或其内部所得迁移在模板下方的部分延伸的引物(***)以及完全延伸的引物(**)。相反,核糖鸟苷复制阻断子几乎能够完全阻止延伸到模板的5'端,仅形成部分延伸的产物。泳道8-11显示了q5du绕路聚合酶在含有r(u)3、r(a)3、r(c)3和r(g)3串的模板上进行引物延伸的反应产物。相较于taq聚合酶,这种酶具有更强的绕过核糖胞嘧啶和核糖鸟苷复制阻断子的能力(泳道10和11),而在包含核糖尿苷和核糖腺苷模板上的延伸,阻断子主要产生部分引物延伸(泳道8和9)。图33b显示了通过kapahifi和gxlprimestar酶在含有不同核糖核苷酸作为复制阻断子的模板上延伸反应产物的电泳分析。泳道2和泳道7显示了延伸引物的迁移,泳道3-6显示了以kapa高保真聚合酶(kapahifi)在含有r(u)3、r(a)3、r(c)3和r(g)3串的模板上引物延伸反应的产物。如泳道3和4所示,kapa高保真酶可以部分地绕过由三核糖尿苷和三核糖腺苷组成的复制阻断子,产生全长以及截短的引物延伸产物。引物延伸反应产生独特条带,对应复制停在复制阻断子开始之处或其内部所得迁移在模板下方的部分延伸的引物(***)以及完全延伸的引物(**)。相反,kapa高保真酶能够完全绕过核糖胞嘧啶和核糖尿苷复制阻断子,仅形成全长延伸产物。泳道8-11显示了以gxlprimestar聚合酶在含有r(u)3、r(a)3、r(c)3和r(g)3串的模板上引物延伸反应的产物。所有四种复制阻断子几乎完全抑制该酶的持续合成能力。图33c显示了通过q5酶在含有不同核糖核苷酸作为复制阻断子的模板上延伸反应产物的电泳分析。泳道2显示了延伸引物的迁移,泳道3-6显示了以q5聚合酶在含有r(u)3、r(a)3、r(c)3和r(g)3串的模板上引物延伸反应的产物。如泳道3至4所示,q5酶不能绕过由三核糖尿苷或三核糖腺苷组成的复制阻断子,仅产生截短的引物延伸产物。相反,q5聚合酶能够完全绕过核糖胞嘧啶和核糖尿苷复制阻断子,仅形成全长延伸产物(泳道5和6)。结论不同的热稳定dna聚合酶具有不同的能力来绕过由3联核糖尿苷、核糖腺苷、核糖胞嘧啶或核糖鸟苷组成的复制阻断子。例如,q5dna聚合酶能够容易地绕过r(c)3和r(g)3复制阻断子,而它在r(u)3、r(a)3序列处完全停滞。相反,相较于q5酶,kapa高保真酶具有更强的通过核糖尿苷和核糖腺苷复制阻断子进行复制的能力。除了非校对性taqdna聚合酶之外所有dna聚合酶都具有共同的性质即不能通过由核糖尿苷组成的复制阻断子进行有效复制,这使得该组成在所测试的四种复制阻断子中成为最通用且具有吸引力的阻断子组成。同时,taqdna聚合酶有无法顺利通过3联核糖g碱基复制问题。实施例4.以标准扩增引物和含有复制阻断子和5'端非互补尾的引物进行的扩增实验的示意图由图34a可知,以标准互补引物一和引物二进行pcr反应可以导致两种不同的情况,所述引物二含有内部复制阻断子和非互补尾。如果dna聚合酶可以绕过复制阻断子,那么模板分子的3'端将延伸至引物2的5'端,产生两条相同大小的dna链。如果dna聚合酶不能通过复制阻断子复制,那么将会产生两条不同大小的dna链,可以在变性凝胶上看到。实施例5.以标准扩增引物和含有复制阻断子和5'端非互补尾的引物使用taq和primestargxldna聚合酶扩增合成的dna模板材料10nm模板寡核苷酸(寡核苷酸11)具有r(u)4复制阻断子和5'端非互补尾部的600nm正向引物(寡核苷酸12)600nm反向引物(寡核苷酸13)taq2x主混合物(neb,目录号m0270l)gxldna聚合酶(克隆泰克公司,cat#r050a)低te缓冲液(天惠华公司目录号to227)25bp梯标dna大小标志物(英杰公司,目录号10488-022)15%tbe-尿素凝胶(英杰公司,目录号ec68852box)sybr金染剂(英杰公司,目录号#s11494)方法采用2xtaq主混合物的扩增反应在50μl反应体积中进行,所述反应体积包含25μl的主混合物,2μl的模板寡核苷酸,2.5μl的扩增引物和18μl的低te缓冲液。采用以下循环参数进行反应:95℃初始酶活化3分钟,然后10轮循环,每轮为95℃下20秒,60℃下30秒和66℃下30秒。采用gxldna聚合酶的扩增在50μl反应体积中进行,所述反应体积包含10μl的5xprimestargxl缓冲液,4μl的dntp混合物(各2.5mm)2μl的模板寡核苷酸,2.5μl的各延伸引物,1μl的primestargxldna聚合酶和28.5μl的低te缓冲液。采用以下循环参数进行扩增反应:98℃初始酶活化30秒,然后10轮循环,每轮为98℃下10秒,60℃下30秒和68℃下30秒。将样品在甲酰胺上样缓冲液中煮沸,20μl各反应在15%tbe-尿素凝胶上200伏特分离。然后凝胶用sybr金染剂染色,在dark读取灯箱(克莱尔化学研究公司)上观察并采用数码相机拍照。结果图34显示了通过taq和gxldna聚合酶在合成模板上以标准引物和含有r(u)4复制阻断子和5'端非互补尾的引物扩增反应产物的电泳分析。泳道2显示了合成寡核苷酸模板的迁移,泳道3显示引物的迁移,泳道4显示了taqdna聚合酶扩增反应的结果,其中顶部条带代表全长扩增产物,而用单个星号标记的少量底部条带显示3'端截短的产物。相反,当使用gxldna聚合酶进行扩增反应时,可以看到两条强度相等的条带,对应于完整大小以及截短的dna。结论如图34b所示,泳道5中,核糖尿苷串可以作为gxldna聚合酶(以及其他几种如q5dna聚合酶,数据未显示)的有效复制阻断子。该现象允许在pcr反应期间由双链dna的一端或两端产生预定义的5'突出端。使用核糖尿苷作为复制阻断子的一个优点是,不同于常规复制阻断子,所产生的转接区可以在后续连接反应中充当底物。实施例6.复杂序列和均聚序列的寡核苷酸结合动力学分析材料500nm复杂底物(寡核苷酸17)500nm复杂探针(寡核苷酸18)500nm均聚底物(寡核苷酸19)500nm均聚探针(寡核苷酸20)2x杂交缓冲液:trisph=7.520mm,mgcl210mm,nacl100mmqubit2.0荧光计(英杰公司目录号q32866)方法杂交反应在200μl体积中进行,其中含有100μl的2x杂交缓冲液,20μl的底物寡核苷酸(50nm终浓度)和10μl荧光探针(25nm终浓度)。使用蓝光激发(430-495nm)和绿光(510-580nm)发射滤光片每1分钟测量一次。结果图35a显示了荧光探针与过量含有淬灭剂的相应底物寡核苷酸的结合速率随时间变化的示意图。荧光探针与底物寡核苷酸的结合导致荧光团非常接近淬灭剂。这导致荧光信号的衰减并且可以通过荧光计测量。虚线显示25nm复杂序列探针与过量互补底物寡核苷酸的结合动力学,而实线显示25nm均聚序列探针与其互补底物的结合(图35b)结论显然,相较于复杂序列探针,在均聚寡核苷酸探针的情况下,与底物的结合速率显著提高。如先前研究所知,两个寡核苷酸的退火涉及形成部分配对的区域及其扩展。当两个复杂序列寡核苷酸在杂交过程中彼此接近时,最初复合体中核苷酸互补的概率非常低。相反,当两个互补的均聚寡核苷酸彼此相遇时,其将导致瞬时成核并形成双链体。这解释了相较于复杂序列探针,均聚探针为何具有显著更快的结合动力学,并且允许在标准化反应中相应的ngs文库标准化探针或引物采用更短地孵育。实施例7.使用含有核酸酶抗性的均聚探针的连接,然后使用外切核酸酶iii的酶促标准化,进行酶法ngs文库标准化材料hapmapdnana12878(科里尔(coriell))accel-2s无pcrdna文库试剂盒(斯威夫特生物科学公司(swiftbiosciences)目录号dl-il2pf-48,si-ilm2s-48a)600nmp5pre-n引物(寡核苷酸14)600nmp7pre-n引物(寡核苷酸15)热启动高保真2x主混合物(neb,目录号m0494s)spri选择dna大小选择珠(贝克曼库尔特公司(beckmancoulter)目录号b23318)低te缓冲液(天惠华公司目录号to227)480nm均聚标准化n-探针(寡核苷酸16)标准化缓冲液n1:trisph=7.562.5mm,mgcl231.25mm,atp6.25mm,dtt62.5mmnacl312.5mm标准化缓冲液n2:trisph=7.510mm,mgcl21mmt4dna连接酶(安赛麦蒂公司(enzymatics)目录号l6030-lc-l)外切核酸酶iii(安赛麦蒂公司目录号x8020f)0.5medtaph=8.0(西格玛-奥德里奇公司(sigma-aldrich)目录号e9884)kapa文库定量试剂盒(kapa生物系统公司目录号kk4824)方法使用来自斯威夫特生物科学公司的accel-2s无pcrdna文库试剂盒由1ngcorielldnana12878构建ngs文库。将文库洗脱在20μl的低te缓冲液中,然后用q5热启动高保真2x主混合物和p5和p7pre-n引物进行10轮循环的扩增。在spri选择dna大小选择珠上使用文库对珠比1.0纯化文库,并用50μl的低tedna再悬浮缓冲液洗脱。使用kapa文库定量试剂盒对扩增所得文库进行定量,并制备一组指定浓度的文库稀释物(图36)。为了将文库稀释物标准化成4nm终浓度,将20μl文库与4μl的n1标准化缓冲液、0.5μl的标准化n-探针和0.5μl的t4dna连接酶混合,并于30℃温育20分钟。然后,将稀释在4.5μl的n2缓冲液中的0.5μl的外切核酸酶iii添加到各管中,并于30℃再孵育20分钟。在孵育结束时,向各管添加1μl的0.5medta,然后95℃加热样品3分钟将酶灭活。根据制造商的说明用基于qpcr的kapa文库定量试剂盒对最终文库浓度进行定量。结果图36a-36b显示标准化之前和之后测序文库的定量结果。对以q5酶扩增的文库进行定量并用低te缓冲液稀释,产生指定浓度在2和100nm之间的20μl等份。然后各等份进行如上所述的文库标准化程序。标准化反应的结果可见于图36a-36b。起始浓度为10nm及以上的所有文库已成功标准化成4nm终浓度,标准偏差为0.21。如所预期,起始物不足的第一个文库不能标准化至4nm终浓度,产生浓度为0.91nm的文库。结论由于限定摩尔量的均聚标准化探针,对不同起始浓度的ngs库进行标准化反应得到具有近似浓度的文库,证明这是一个测序之前简单、可靠的ngs库标准化方法。实施例8.使用具有荧光素染料和淬灭剂基团的寡核苷酸的再结合动力学显示具有重复尾的引物退火快约10倍材料500nm常规底物(寡核苷酸21)500nmgt加尾的底物(寡核苷酸22)500nmac加尾的引物(寡核苷酸23)2x杂交缓冲液:trisph=7.520mm,mgcl210mm,nacl100mm协同htx多模式酶标仪(伯腾仪器公司(biotek))方法杂交反应在100μl体积中进行,其中含有50μl的2x杂交缓冲液,10μl的底物寡核苷酸(50nm终浓度)和5μl荧光探针(25nm终浓度)。使用蓝光激发和绿光发射滤光片每1分钟测量一次。结果图37a显示了荧光探针与过量的两种不同寡核苷酸底物的结合速率随时间的变化。荧光探针与底物寡核苷酸的结合导致荧光团非常接近淬灭剂。这导致荧光信号的衰减并且可以通过荧光计测量,图37。虚线显示25nm探针与过量的常规模板寡核苷酸的结合动力学,其中固体泳道显示了探针与含有gt重复的模板寡核苷酸的结合(参见图37b-37c探针和寡核苷酸对的示意图)。结论显而易见的是,相较于常规复杂模板,在含有gt重复的模板的情况中,与底物的结合速率显著地增加。如先前研究所知,两种寡核苷酸的退火涉及形成部分配对的区域及其扩增。先前已经显示,两个寡核苷酸的退火涉及形成部分配对的区域及其延伸。当两个复杂寡核苷酸在杂交过程中彼此接近时,初始复杂序列中核苷酸互补的概率非常低。相反,当含有gt和ac重复的两个寡核苷酸彼此相遇时,其将导致更快的成核和双链体形成。这解释了相较于常规模板,探针与含有gt重复的模板更快的结合动力学,并且能够在标准化pcr反应中允许标准化引物和ngs文库更短的退火时间。实施例9.使用基于pcr的标准化(n-pcr引物)进行8个accel-ngs2s文库的标准化材料400nm常规pcr引物1(寡核苷酸24)400nm常规pcr引物2(寡核苷酸25)400nmn-pcr引物1(寡核苷酸26)400nmn-pcr引物2(寡核苷酸27)hapmapdnana12878(科里尔)accel-2s无pcrdna文库试剂盒(斯威夫特生物科学公司目录号dl-il2pf-48,si-ilm2s-48a)热启动高保真2x主混合物(neb,目录号m0494s)低te缓冲液(天惠华公司目录号to227)kapa文库定量试剂盒(kapa生物系统公司目录号kk4824)方法使用来自斯威夫特生物科学公司的accel-2s无pcrdna文库试剂盒由100ngcorielldnana12878构建测序文库。将文库洗脱在20μl的低te缓冲液中,并使用kapa文库定量试剂盒对文库进行定量,然后制备一组指定浓度的文库稀释物(图38a-38b)。为了将文库稀释物标准化至40nm最终浓度,对文库进行标准化pcr反应,其中包含15μl的文库稀释物、25μl的热启动高保真2x主混合物和5μl的各种常规或n-pcr引物(引物浓度为40nm)。采用以下循环参数扩增文库:98℃初始酶活化45秒,然后4轮循环,每轮为98℃下10秒,60℃下5分钟,72℃下1分钟,然后18轮循环,每轮为98℃下10秒,60℃下1分钟,72℃下1分钟。使用kapa文库定量试剂盒对扩增的文库进行定量。结果图38a-38d显示标准化之前和之后测序文库的定量结果。对ngsdna文库进行定量并用低te缓冲液稀释,产生指定浓度在1000和7.8pm之间的15μl等份。然后各等份进行如上所述的文库标准化pcr。以n-pcr引物扩增的所有文库被成功标准化至大致相同的浓度。相反,以常规引物对相同样品进行标准化扩增显示近乎100%的变差。结论使用n-pcr引物对8个accel-ngs2s文库进行标准化显示约10%的相对偏差,其中2s文库输入差异超过100倍。对于相同的样品,常规引物显示近乎100%的变差,证明当pcr引物浓度在反应中限制pcr产物得率时,相比常规引物使用n-pcr引物扩增指定量pcr产物的优势。实施例10.使用基于pcr的标准化(n-pcr引物)进行accel-扩增子靶向的ngs文库的标准化材料400nmn-pcr引物1(寡核苷酸26)400nmn-pcr引物2(寡核苷酸27)hapmapdnana12878(科里尔)accel-扩增子56g肿瘤组+样品_id(斯威夫特生物科学目录号al-56248)热启动高保真2x主混合物(neb,目录号m0494s)低te缓冲液(天惠华公司目录号to227)kapa文库定量试剂盒(kapa生物系统公司目录号kk4824)方法根据生产商说明,使用来自斯威夫特生物科学公司的accel-扩增子56g肿瘤组试剂盒由10ngcorielldnana12878构建扩增子文库。将文库洗脱在20μl的低te缓冲液中,并使用kapa文库定量试剂盒对文库进行定量,然后制备一组指定浓度的文库稀释物(图39)。为了将文库稀释物标准化至40nm最终浓度,对文库进行标准化pcr反应,其中包含15μl的文库稀释物、25μl的热启动高保真2x主混合物和5μl的各种n-pcr引物(引物浓度为40nm)。采用以下循环参数扩增文库:98℃初始酶活化45秒,然后4轮循环,每轮为98℃下10秒,60℃下5分钟,72℃下1分钟,然后16轮循环,每轮为98℃下10秒,60℃下1分钟,72℃下1分钟。使用kapa文库定量试剂盒对扩增的文库进行定量。然后将文库稀释10倍,汇集在一起并以20pm加载浓度使用miseq试剂纳米盒(reagentnanokit)v2在miseq测序仪(illumina)上测序。使用illumina测序分析观测软件(illuminasequencinganalysisviewersoftware)计算集合中各文库在流动池上可得成簇%。结果根据试剂盒方案构建56g扩增子文库,定量并以低te缓冲液稀释180、60、30和18倍。如上所述,然后对文库进行标准化pcr,定量和测序。如图39a-39c所示,所有8个文库都成功标准化至预期的40nm浓度。测序结果还证明了各文库在流动池上形成预测%的簇,等于约12.5%。结论基于pcr的ngs库标准化是标准方法的简单替代方案以简化制备加载测序仪所用文库的工作流程。对各文库实现的文库得率与n-pcr引物浓度成比例,由此生成的文库集合中文库在illumina流动池上显示相等负载。实施例11.使用n-pcr引物进行16个illumina文库的标准化证明了使用基于qpcr和基于荧光的定量方法的相对定量准确性材料400nm荧光标记的n-pcr引物1(寡核苷酸28)400nmn-pcr引物2(寡核苷酸29)200nm淬灭剂寡核苷酸(寡核苷酸30)由合作者提供的16个illumina库热启动高保真2x主混合物(neb,目录号m0494s)低te缓冲液(天惠华公司目录号to227)kapa文库定量试剂盒(kapa生物系统公司目录号kk4824)协同htx多模式酶标仪(伯腾仪器公司)方法使用kapa文库定量试剂盒对illumina文库进行定量并稀释20倍。为了将文库稀释物标准化至40nm最终浓度,对文库进行标准化pcr反应,反应包含2μl的文库稀释物,25μl的热启动高保真2x主混合物和5μl的各种引物(最终浓度40nm)和13μl的低te缓冲液。用以下循环参数扩增文库:98℃初始酶活化45秒;然后4轮循环,每轮为98℃下10秒,60℃下5分钟,72℃下1分钟;然后7轮循环,每轮为98℃下10秒,60℃下1分钟,72℃下1分钟。使用kapa文库定量试剂盒或协同htx多模式酶标仪对扩增所得文库进行定量。结果实施例11的结果显示qpcr标准化文库的定量结果(图40a-40d)。如图所示,以n-pcr引物扩增的所有文库成功地标准化至大致相同的浓度。协同htx多模式酶标仪荧光试验获得了更高一致性的相对定量。添加猝灭寡核苷酸后整体荧光水平下降约10%。该观测结果反映了这样的事实:并非所有的标准化引物都被标准化pcr反应所利用,其中10%的下降对应约10%未被纳入的标记的引物。结论以上数据表明,利用荧光标记的引物结合淬灭寡核苷酸的荧光试验可以是用于文库定量的快速且可靠的方法,并且不需要将文库与未使用的引物纯化分离,并且在文库插入大小未知或者插入大小分布较宽的情况中,不需要关于文库插入大小的信息来计算摩尔浓度。实施例12.包括标准化衔接子-探针与带截短衔接子序列文库连接的通过合成进行illuminangs文库的标准化该实施例证明如图42e和42g所概述的合成法ngs文库标准化的可行性,并且显示了文库标准化可以单个酶促步骤中完成,即通过将指定摩尔量的双链或单链标准化探针与dna文库(t-文库)一端的截短ngs衔接子连接产生等量的功能性ngs文库。材料斯威夫特生物科学公司accel-2s无pcrdna文库试剂盒,目录号200246um引物p7(寡核苷酸31)6um引物pt1(寡核苷酸32)100nm双链探针nt1(由寡核苷酸33和34的退火形成)6um引物pt2(寡核苷酸35)100nm单链探针nt2(寡核苷酸36)热启动高保真2x主混合物(neb,目录号m0494s)t4dna连接酶,120,000u/ml(安赛麦蒂公司目录号l6030-lc-l)10xt4dna连接酶缓冲液(安赛麦蒂公司目录号b6030l)低te缓冲液(天惠华公司目录号to227)hapmap人dna(科里尔生物库(coriellbiorepository),na12878)invitrogenqubit2.0荧光计,目录号q32866qubitdsdnabr分析试剂盒,目录号q32853kapa文库quantillumina试剂盒,目录号kk4824spri选择dna大小选择珠(贝克曼库尔特公司目录号b23318)方法使用斯威夫特生物科学公司accel-2s无pcrdna文库试剂盒由人coriellna12878dna构建ngs文库。如图43a和43b所示,使用引物p7和引物pt1或引物pt2通过pcr扩增文库。pcr反应体积为50ul,其中含有25ul的q52x高保真主混合物,5ul的寡核苷酸31(600nm),5ul的寡核苷酸32(600nm),5ul经1:500稀释的2sngs文库和10ul低te(10mmtris,0.1mmedta)。扩增条件是:98℃下45秒,然后是每轮98℃下10秒和66℃下1分钟的30轮循环。完成pcr反应后,按照dna:珠比1.0在spri珠上纯化文库,用50μl的低tedna缓冲液洗脱,并使用invitrogenqubit2.0荧光计和与之配合的qubitdsdnabr分析试剂盒进行定量。200nm、150nm、100nm、50nm和25nm浓度的经稀释截短文库(t-文库)与120单位的t4dna连接酶和10nm标准化n-探针在t4dna连接酶缓冲液中30℃孵育1小时。以引物pt1扩增的t文库与双链标准化探针nt1孵育,而以引物pt2扩增的t文库与单链探针nt2孵育。为了测定产生的功能性ngs文库的摩尔浓度,以kapaqpcr法文库quantillumina试剂盒对连接产物进行定量。结果实验工作流程如图43a所示,标准化过程和相应的引物、衔接子和探针序列如图43b和43c所示。10nm双链标准化探针nt1连接(图43b)或10nm单链探针nt2连接(图43c)产生全尺寸文库,其中截短衔接子(p5)复原为全长成为功能性ngs文库。通过qpcr定量评估文库合成和标准化的完成度。如图43d所示,虽然t文库的输入量为200到25不等,但生成文库的摩尔浓度都在预期的10nm值量级且都在所用定量方法的精度(约20%)之内。虽然在本研究中使用的原始文库是全尺寸ngs库,但是其被稀释500倍后用引物pt1或pt2扩增以产生截短t文库,因此原始文库对qpcr定量的预期贡献可忽略不计。结论以上提供的数据提供了这样的证据,即通过受控合成进行ngs文库标准化是简单且稳健的,并且可以用于测序之前的文库制备,以代替耗时耗力的文库定量和浓度调整。实施例13.使用双链dna部分特异性荧光染剂的合成法标准化的ngs文库的定量实施例12以及本文别处所述的通过合成进行文库标准化产生双链功能性ngs文库,并且理论上可以用双链dna特异性荧光染料对其进行染色。不幸的是,剩余的非功能性文库部分也是双链的,也可能被染色,结果导致无法清晰定量完整的功能性文库部分。这个问题可以通过在定量之前以外切核酸酶iii孵育文库来解决。图14显示该过程的详细描述。在所示方法中,以含有多聚ru或多聚ra复制挡块的pcr引物扩增一个衔接子端截短的ngs文库,如本公开其他部分所述。具体地,引物之一包含与实施例12中pt1引物中相同的截短衔接子序列,而p7引物另外具有含有多聚(ru)或多聚(ra)碱基的5'尾。以高保真dna聚合酶如q5、primestargxl或kapahifi进行的pcr扩增所得文库分子在两个衔接子端具有5'突出端。当截短p5衔接子末端的突出序列由p5衔接子序列决定时,可以选择完整p7衔接子端的突出序列,使其没有与标准化探针退火的互补性。除了在其3'末端还具有一个或多个核酸酶抗性碱基的标准化探针(不参与连接反应)之外,标准化反应混合物还含有保护帽分子。保护帽分子被设计成具有与p7衔接子端的5'突出端互补的5'突出端(在pcr期间产生),具有核酸酶抗性并且摩尔量过量,由此为所有p7衔接子末端提供抗外切核酸酶iii的保护。保护帽短小且不太可能在illumina设备上簇形成和/或测序过程期间影响文库性能。在两个3'端都含有核酸酶抗性碱基的标准化文库部分对外切核酸酶iii处理具有抗性并且保持双链。另一方面,仅在一个衔接子端具有核酸酶抗性碱基的所有截短文库分子转变成单链形式,不能用sybr或qubit等双链特异性染剂检测到。在没有保护帽分子的情况下,该方案是不可行的,因为在这种情况下,包括标准化部分在内的所有文库分子都将被转化为不可染色的单链形式。实施例14.合成长范围扩增子文库用于单链测序单分子测序是一种非常有前景且快速发展的ngs种类,其可以对非常长的dna分子进行序列分析。与illumina和iontorrent测序平台的情况一样,单分子测序也需要将dna片段转化成在一端或两端携带专门的衔接子序列的平台特异性ngs文库。在太平洋生物科学公司(pacificbiosciences)平台的情况下,衔接子是位于两端的单一茎-环,而在牛津纳米孔公司(oxfordnanopore)平台的情况下,它是共价连接蛋白质分子的y形dna结构。通常,测序过程涉及并行dna样品,另外还使用插入衔接子和插入dna序列之间的样品id序列(标引或条码)。当前衔接子和样品标引方法耗时和/或需要多轮spri珠纯化以除去未连接的衔接子分子。我们在此提出了一个简单的方案,其能够在pcr期间连接茎-环衔接子序列,因此在测序之前仅需要断口(nick)闭合或缺口(gap)填平反应(图45-47)。缺口填平反应也可用于插入用于并行应用的标引序列。通过用修饰特异性内切核酸酶切割环可以很容易地将茎环衔接子转化为y形衔接子。材料6um引物(寡核苷酸37)6um引物(寡核苷酸38)10μm线性通用引物(寡核苷酸39)10μm茎-环通用引物(寡核苷酸40)10μm茎-环衔接子(寡核苷酸41)1μm标引接头(寡核苷酸42)大肠杆菌migula基因组dna(atcc,目录号mg1655)spri选择dna大小选择珠(贝克曼库尔特公司目录号b23318)t4dna连接酶,120,000u/ml(安赛麦蒂公司目录号l6030-lc-l)10xt4dna连接酶缓冲液(安赛麦蒂公司目录号b6030l)低te缓冲液(天惠华公司,目录号to227)外切核酸酶iii,100,000u/ml(安赛麦蒂公司目录号x8020f)外切核酸酶vii,10,000u/ml(neb目录号mo263s)热启动高保真2x主混合物(neb,目录号m0494s)2100生物分析仪(bioanalyzer)(安捷伦(agilent),目录号g2939ba)高灵敏度dna芯片(安捷伦,目录号5067-4626)方法通过pcr反应使用加尾的引物(寡核苷酸37和38在50ul体积中产生一级大肠杆菌扩增子,所述体积包含25ul的q52x高保真主混合物,5ul的寡核苷酸37,5ul的寡核苷酸38,5ul的大肠杆菌dna储液和10ul的低te缓冲液。扩增条件是:98℃下45秒,然后每轮98℃下10秒和66℃下1分钟共进行26轮循环。完成pcr反应后,将文库在spri珠上使用dna:珠比1.2纯化,并用50μl低te缓冲液洗脱。以下述条件使用通用线性引物(寡核苷酸39)或通用茎-环引物(寡核苷酸40)再次扩增一级扩增子产物:5ul100倍稀释的第一pcr反应所得扩增子dna,25ul的q52x高保真主混合物,5ul的通用引物和15ul的低te缓冲液,最终体积为50ul。扩增条件是:98℃下45秒,然后每轮98℃下10秒和66℃下1分钟共进行30轮循环。将pcr产物在spri珠上使用dna:珠比1.2纯化,并用20μl低te缓冲液洗脱。通过用t4dna聚合酶以下述条件孵育线性通用引物的pcr产物与茎-环衔接子(寡核苷酸41)和标引接头寡核苷酸(寡核苷酸42),以及茎-环通用引物的pcr产物与标引接头寡核苷酸(寡核苷酸42),进行衔接子连接和缺口填平反应(线性通用引物,图45a)或仅进行缺口填平反应(茎-环通用引物,图45b):3ul的pcr产物,5ul的寡核苷酸41,10ul的寡核苷酸42,4ul的10xt4连接缓冲液,2ul的t4连接酶和低te缓冲液,以40ul的总反应体积。连接反应在30℃进行30分钟,然后将一半反应系用外切核酸酶iii/外切核酸酶vii混合物另外处理15分钟。外切核酸酶处理后,在spri珠上使用文库:珠比1.2纯化反应系,并使用高灵敏度dna芯片在安捷伦2100生物分析仪上解析。结果在第二pcr反应中使用含有(核糖u)4的线性(图45a)或茎-环(图45b)通用引物产生这样的dna分子,其具有含有序列(gt)6(t)22(ru)4的单链5'突出端(图45a)或含有序列(t)22(ru)4的单链缺口区域和茎-环突出端(图45b)。在第一种情况中的单链尾和在第二种情况中的单链缺口成为茎-环衔接子和标引接头(第一种情况)或只是标引接头(第二种情况)退火和连接的模板。图45显示生物分析仪数据表明,线性引物pcr后与的衔接子/接头连接的产物(泳道5)和茎-环引物pcr后的接头连接产物(泳道7)具有对外切核酸酶iii/外切酶vii联合处理的抗性(相应地为泳道6和8),这表明文库末端具有预期的哑铃状结构。数据显示,超过50%的pcr扩增子转化为外切核酸酶抗性分子。泳道1、2、3和4分别显示了dna梯标、第一pcr反应产物和上述两种第二pcr反应的产物。结论提供的数据描述了一种新的高效方法,用于制备在两端具有茎-环衔接子的扩增子文库。所该方法还包括通过将id序列包含在接头寡核苷酸中来纳入样品id序列的有效方式,因此不需要合成长条码化的接头寡核苷酸。通过将经修饰的可切割碱基(如du,rna,脱氧肌苷,甲基化胞嘧啶等)导入pcr引物的环区域并通过修饰特异性内切核酸酶(如user酶混合物,rna酶,内切核酸酶v,甲基化特异性内切核酸酶等)进行切割,茎-环引物法可以无限制地用于生成在末端具有y形衔接子的扩增子文库。这样的切割可与标引/条码接头连接步骤组合,将整个过程限于单个孵育反应。如图46a和46b所示,实施例14中所示方法不限于接头寡核苷酸连接产生共价闭合的dna结。该方法优选但不限于在5'突出端使用均聚物或/和二核苷酸重复序列以实现茎-环或y形衔接子连接,如图47a所示,其中5'突出序列x可以是任何dna序列。根据本公开提供的数据,当与高保真dna聚合酶组合使用时,三个或更多个碱基的ru和ra串将提供最佳复制终止。另一方面,三个或更多个rg碱基为taqdna聚合酶提供了复制终止。茎-环衔接子连接还可以利用通过t4dna聚合酶产生的5'突出端,如图47b所示。在这种情况中,pcr引物产生扩增子并纳入含有两种不同dna序列的尾部:低复杂性序列x和非x区中碱基的缓冲序列b。例如,序列x是均聚物aaa...aaa,序列b仅含有g和c碱基。在存在t4dna聚合酶和含有缓冲区核苷酸(例如,dgtp和dctp)但不存在与x区互补碱基(dttp)的限定核苷酸混合物的情况下,pcr扩增子的末端将被修剪以产生x突出端(多聚a尾)。可以在t4dna连接酶加热灭活后连接具有与尾序列x互补的序列x'的茎环衔接子。或者,在茎-环衔接子具有碱基组成相似但不一定为相同序列的缓冲序列b的情况下,连接反应可以与t4dna聚合酶修剪反应组合。缓冲区阻止t4dna聚合酶介导的外切核酸酶消化茎环衔接子(像它作用于扩增子末端那样)。实施例15.长片段扩增子的寡聚化,用于更有效的单分子(太平洋生物科学公司和牛津纳米孔公司)测序开发的用于单分子测序的方法能够对长达50(pacbio)或甚至800(ont)千碱基的非常长的dna序列进行测序,如太平洋生物科学公司方法,该方法通过固定的phi29dna聚合酶在引物延伸反应期间连续检测纳入dna中的荧光碱基,或如牛津纳米孔公司方法,该方法在通过纳米孔进行dna扩增期间检测序列特异性电流波动。当通过这类设备分析明显更短的dna分子时,它们的利用效率显著降低。为了克服这个问题,可以将dna分子连接到更长的多联体结构中。dna寡聚化也可用于克服芯片负载中的尺寸偏差,这对pacbio仪器来说尤为明显。如果进行共同测序,短dna分子和长dna分子不同的负载效率使得对大小分布在1至10kb之间(具有约2kb的峰)不同大小cdna分子的表达分析产生显著的大小偏性。本申请所述用于有效产生5'突出端的方法可用于产生多联体扩增子分子。我们设想了用于扩增子寡聚化的多种策略。在一种策略中,如图48a、48c和48d所示,单独进行pcr(用含有核糖u的引物,图48a、48c)或pcr并随后用t4dna聚合酶孵育(图48a、48d)产生具有互补5'突出端a和a'的dna分子,类似于实施例14中所述产生用于茎-环衔接子连接的这类突出端。在dna连接酶存在的情况下,具有互补突出端的dna分子将产生长共价多联体,类似于在末端具有互补的12个碱基的突出端的完整噬菌体λdna的寡聚化。在另一种策略中,如图48b所示,单独进行pcr(用含有核糖u的引物)或pcr并随后用t4dna聚合酶孵育产生具有非互补5'突出端a和b的dna分子。在这种情况中,通过将扩增子与化学计量等摩尔量的双链接头dna混合来实现dna寡聚化,所述双链接头dna含有突出端a'和b'。在另一种策略中,以含核糖u的引物进行pcr扩增将产生具有部分互补突出端的扩增子,如图48e所示。在这种情况中,仅有两个5'突出端的5'部分互补,并具有gtgtgtgtgtgt和acacacacacac序列。扩增子的退火产生非共价结合的寡聚体,其中含有位于ru碱基旁边含均聚物多聚t序列的单链缺口区。含有多聚a均聚物序列的接头寡核苷酸的添加和连接形成共价寡聚物。接头寡核苷酸不仅可用于完成扩增子寡聚体的形成,还可用于纳入标引序列用于样品并行处理。实施例16.产生可再扩增的单链探针用于基于杂交-捕获的靶标富集对于制备这样的单链分子存在很高的需求,所述单链分子仅含有pcr扩增所得dna中的选定链。由双链pcr产物制备单链dna是用“通过指数富集进行配体系统性进化(selex)”鉴定适体的必要步骤。其还经常用于基因分型和基于dna的诊断检测。有些方法利用λ5'外切核酸酶消化含有5'磷酸基团的dna链,同时保留未磷酸化的链。不幸的是,λ外切核酸酶对磷酸化dna末端的特异性不是绝对的,这导致未磷酸化dna链的非特异性降解。其他方法使用在链霉亲和素磁珠上固定含有生物素的pcr产物,并且通过naoh处理以及酸中和从珠中选择性释放感兴趣的链。该方法不适用于制备生物素化的dna或任何大规模单链dna制备。这里我们提出了一种新型方法,用于大规模生成单链dna分子,特别是来自pcr产物的生物素化单链分子。所提出的方法是高效的,具有低生产成本并且可扩展至大体积和探针数量,因此,理想地适用于制备杂交捕获探针用于定向富集dna和rna供ngs分析之用。该方法还可以用于生产用其他配体或发色团标记的单链dna。该方法是链特异性的,并且允许制备针对两条dna链的探针。该方法包括多个步骤(图49a)。首先,使用含有通用序列a和b的引物以及热稳定dna聚合酶pcr扩增来自基因组或载体dna的选定区域。第二步,以通用引物a和b和高保真dna聚合酶如q5、primestargxl或kapahifi将pcr产物稀释和再扩增,其中引物a具有生物素基团,而引物b具有位于5'端的10-16dt碱基串之后的(ru)4复制挡块。pcr产生一端具有5'突出端的双链dna分子。还可以使用在其5'端具有(ru)4(dt)12-16序列以及生物素的靶标特异性引物在一个pcr步骤中产生具有5'突出端的靶dna分子。用于复制终止的核糖核苷酸碱基的选择不仅限于ru碱基,其可以是多聚(ra)或任何其他核糖核苷酸序列只要它们能够有效阻止为扩增所选dna聚合酶的聚合反应并产生足以用于退火和连接核酸酶抗性寡核苷酸的5'突出端。第三步,退火和连接含有10-20个a碱基、5'磷酸基团和5'端附近至少一个硫代磷酸酯键的核酸酶抗性寡核苷酸。优选地,存在至少4个硫代磷酸酯键以更好地提供抗外切核酸酶保护。引物b的5'尾序列和核酸酶抗性寡核苷酸c的互补序列不限于多聚t和多聚a序列,可以代之以任何其他彼此互补的序列。如实施例6所示,选择多聚t和多聚a序列的理由在于其快速的退火和连接时间。第四步,用ampurexp珠进行纯化,以去除未纳入的pcr引物和保护寡核苷酸。寡核苷酸c的量可以过量,由此保护pcr反应产生的所有dna分子,或者如果目标是仅产生指定量的单链探针,那么寡核苷酸c的量是限定的,以指定的摩尔浓度存在。第五步,用外切核酸酶iii消化以去除未保护的dna链。最后,通过旋转柱或任何其他可用的方法进行探针纯化或外切核酸酶iii热灭活。以上所述方法产生生物素化的,单链dna捕获探针,其链特异性取决于通用引物上的(ru)4(dt)12-16序列和生物素基团的位置。通过将生物素基团移至引物b,并相应地将(ru)4(dt)12-16序列移至引物a可以产生与第二dna链互补的捕获探针(图49b)。可以将生物素化的链特异性探针汇集成组用于通过与变性ngs库杂交来分离多个目标区域。为确保探针以相同浓度存在,应当使用标准方法对其量进行定量并通过稀释调整浓度。如果已经通过受控连接核酸酶抗性探针c对探针量进行了标准化,那么多探针汇集就能得以简化。在这样的情况中就可以省略汇集前的探针浓度测定和调整。以上所述方法提供了一种用于探针生成的无限资源,因为如图49c所示,汇集的dna探针可以通过pcr以两个通用引物重复扩增,与核酸酶抗性寡核苷酸连接,并用外切核酸酶iii消化,由此产生单链、链特异性、含生物素的dna捕获探针。实施例17t4dna聚合酶介导突出端生成对缓冲区的要求本实施例数据显示,由最稳定的g和c碱基组成的缓冲dna区域应当至少4个碱基长以防止t4dna聚合酶进行不可逆的dna末端修剪。材料寡核苷酸48寡核苷酸49寡核苷酸50寡核苷酸51寡核苷酸52寡核苷酸53寡核苷酸54寡核苷酸5510xt4dna连接酶缓冲液(安赛麦蒂公司目录号b6030l)低te缓冲液(天惠华公司目录号to227)t4dna聚合酶(neb,30000u/ml,目录号m0203l)dgtp,100mm(目录号55084)dgtp,100mm(目录号55084)15%tbe-尿素凝胶(英杰公司,目录号ec68852box)sybr金染剂(英杰公司,目录号#s11494)方法通过5□m寡核苷酸48与5□m寡核苷酸49,5□m寡核苷酸50与5□m寡核苷酸51,5□m寡核苷酸52与5□m寡核苷酸53和5□m寡核苷酸54与5□m寡核苷酸55在50mmnacl中25℃退火30分钟制备图50中的双链寡核苷酸构建体a、b、c和d,它们具有位于dna一端的8碱基长的缓冲gc区,位于中间的不同长度的缓冲gc区域和位于dna另一端的多聚t/多聚a序列。将样品在甲酰胺上样缓冲液中煮沸,15%tbe-尿素凝胶上200伏特分离。凝胶用sybr金染剂染色,在dark读取灯箱(克莱尔化学研究公司)上观察并采用数码相机拍照。结果图50a-b显示了以t4dna聚合酶孵育之前和之后寡核苷酸构建体和凝胶变性凝胶分析的结果。前4个泳道显示了在与t4dna聚合酶孵育之前构成构建体a-b的寡核苷酸(寡核苷酸48-55)的移动性。互补寡核苷酸迁移为单一条带,因为它们具有相似的大小。在dgtp和dctp核苷酸存在下用t4dna聚合酶处理后,出现了新的、迁移更快的条带,其对应被t4dna聚合酶修剪的多聚a序列。由于构建体另一端上的9个碱基ggcggcggc缓冲序列,第二条链保持完整。分别含有缓冲区ggcggc和ggcg的构建体a和b产生强烈的电泳带,指示在缓冲区处显著的聚合酶停止。含有2碱基缓冲区gg的构建体c未在预期位置停止t4dna聚合酶作用,并产生对应于位于gg位点下游的第二、3碱基缓冲序列gcg的较短扩散电泳条带。有趣的是,含有单个g碱基作为缓冲区的构建体d不能令t4dna聚合酶停在内部3碱基序列gcg处。该结果表明,观察到的构建体c的扩散条带表示的是用t4dna聚合酶孵育15分钟后检测到的瞬时中间状态,这是由于存在由gg二核苷酸提供的上游修剪阻挡。结论本实施例数据表明,4个g/c碱基代表缓冲区阻止dna聚合酶修剪通过缓冲区的最小尺寸。3个g/c碱基是不足的,仅能临时阻止t4dna聚合酶的3'外切核酸酶活性。该结论对于g/c缓冲组成是成立的,但如果t和a碱基用作缓冲区则可能需要更长的尺寸。由于t4dna聚合酶较高的外切核酸酶活性,在较高温度下修剪可能需要较长的缓冲区。这样的要求与本领域已知的\以t4dna聚合酶进行非连接依赖性克隆显著不同,那其中,通常用单胞嘧啶碱基和限定核苷酸混合物由t4dna聚合酶产生5'突出端。我们的数据表明,对于需要精确的5'突出长度和序列的应用,建议使用5个或更多缓冲g/c碱基、也可能6个或更多a/t碱基,以确保产生可预测的dna突出端结构因此,本发明非常适于实现本文所述以及其中隐含的固有的目的和优点。本领域技术人员可作出许多本发明构思范围(部分地由所附权利要求表明)内的改变。本公开特定实施例的以上描述是为了阐述和说明的目的。对这些示例性的实施方式进行选择和描述是为了更好地解释本文所述原理及其实际应用,以使本领域其它技术人员能够更好地利用本主题以及具有适用于预期的特定用途的各种改良的各种实施方式。本公内的各种实施方式的不同特征和公开内容可以在本公开的范围内组合。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1