包含促进剂的热固化环氧树脂组合物的制作方法
本发明涉及抗冲热固化环氧树脂组合物领域,所述环氧树脂组合物特别用作车身粘合剂并且用于制备结构泡沫。
背景技术:
热固化环氧树脂组合物长期已知。一段时间以来人们已经努力消除或至少大大减少环氧树脂组合物的巨大缺点,即其脆性,脆性在冲击应力下会导致经固化环氧树脂组合物出现裂纹或破坏。已经通过使用韧性改性剂或通过环氧树脂的化学改性进行尝试。
热固化环氧树脂组合物的一个重要的应用领域是车辆制造,特别是车身中空腔的粘合或发泡填充。在两种情况下,在施用环氧树脂组合物之后在ktl-(阴极浸涂)-炉中加热车身,使得热固化环氧树脂组合物固化并且任选发泡。
为了能够进行迅速固化,除了用于环氧树脂的可热活化固化剂之外还可以使用促进剂。已知类别的促进剂例如为脲、潜在的咪唑以及胺-三氟化硼复合物。
然而目前市场上正在努力降低ktl-炉的温度。因此市场上非常需要即使在较低温(即130至140℃的温度)下在较短时间(通常10至15分钟)之后就已经固化的热固化环氧树脂组合物。当为此例如使用芳族脲时(芳族脲由于其结构而非常具有反应性),会造成热固化环氧树脂组合物的储存稳定性的严重问题。因此需要一方面在较低温下固化但另一方面具有足够储存稳定性的热固化环氧树脂组合物。
发明概述
因此本发明的目的是提供一种抗冲热固化环氧树脂组合物,所述环氧树脂组合物一方面在室温下具有良好的储存稳定性,另一方面在130℃至140℃的温度下能够迅速固化。
出人意料地,通过根据权利要求1所述的热固化环氧树脂组合物实现该目的。所述环氧树脂组合物特别适合用作单组份热固化粘合剂,特别在车辆制造中用作热固化单组份车身粘合剂,以及用于制备结构泡沫以增强空腔,特别是金属结构的空腔。
本发明的其它方面为其它独立权利要求的主题。本发明的特别优选的实施方案为从属权利要求的主题。
发明详述
本发明涉及单组份热固化环氧树脂组合物,其包含
a)至少一种每分子具有平均多于一个环氧基团的环氧树脂a;
b)双氰胺b;
c)至少一种促进剂c,其中所述促进剂c为通过如下物质的反应获得的化合物:
-1重量份的胺/环氧-加合物(a),和
-0.1至0.8、优选0.2至0.6重量份的化合物(b),所述化合物(b)选自酚树脂和多元酚化合物;
其中胺/环氧-加合物(a)通过如下物质和至少一种每个分子具有平均多于一个环氧基团的环氧树脂(3)的反应获得:
-下式的氨基化合物(1)
其中r3和r4彼此独立地各自表示具有1至5个碳原子、特别是1至2个碳原子、特别优选2个碳原子的烷基,并且r5表示具有1至5个碳原子、特别是3个碳原子的亚烷基;或
-优选氨基化合物混合物,所述氨基化合物混合物以70/30至99/1、特别是80/20至97/3的(1)与(2)的重量比包含氨基化合物(1)和1-氨基-4-乙基哌嗪(2);
其中两者比例使得对于氨基化合物(1)或(1)+(2)中的每当量氨基,环氧树脂(3)中的环氧基团的量为0.8至2.5、特别是1.0至1.6当量;
d)至少一种增韧剂d;
其中以克/摩尔环氧树脂a的环氧基团计,双氰胺b的比例为3-40g/mol环氧基团,并且
其中以克/摩尔环氧树脂a的环氧基团计,促进剂c的比例为5-35g/mol环氧基团。
在本文中,关于取代基、基或基团使用表述“彼此独立地”表示:在相同分子中以相同方式标明的取代基、基或基团可以同时以不同含义出现。
“增韧剂”在本文中被理解为环氧树脂基质的添加剂,以环氧树脂组合物的总重量计,所述添加剂在≥5重量%,特别是≥10重量%的低加入量下造成韧性明显增加,因此能够在基质撕裂或断裂之前获得更高的弯曲应力、拉伸应力、冲击应力或碰撞应力。
物质名称(例如“多元醇”、“多异氰酸酯”、“聚醚”或“多胺”)中的前缀“聚/多”在本文中表示,每种物质在形式上每分子包含多于一个在其名称中出现的官能团。
“分子量”在本文中被理解为分子的摩尔质量(以克/摩尔计)。“平均分子量”表示低聚或聚合形式的分子混合物的数均分子量mn,其通常通过gpc相对于聚苯乙烯标样确定。
“伯羟基”表示结合至具有两个氢的c原子上的oh-基团。
术语“伯氨基”在本文中表示结合至一个有机基团上的nh2-基团,而术语“仲氨基”表示结合至两个有机基团(它们也可以是环的公共部分)的nh-基团。因此具有伯氨基的胺被称为“伯胺”,具有仲氨基的胺被称为“仲胺”,并且具有叔氨基的胺被称为“叔胺”。
“室温”在本文中表示23℃的温度。
每分子具有平均多于一个环氧基团的环氧树脂a优选为液体环氧树脂或固体环氧树脂。术语“固体环氧树脂”是环氧化物领域的技术人员熟知的并且与“液体环氧树脂”对照使用。固体树脂的玻璃化转变温度高于室温,即其在室温下可以粉碎成能自由流动的粉末。
优选地,环氧树脂具有式(ii)
在此,取代基r'和r”彼此独立地表示h或ch3。
对于固体环氧树脂,指数s表示>1.5,特别是2至12的值。
这种固体环氧树脂例如可从dow或huntsman或hexion市售获得。
指数s在1和1.5之间的式(ii)的化合物被本领域技术人员称为半固体环氧树脂。所述半固体环氧树脂对于本发明而言同样被视为是固体树脂。然而优选狭义上的固体环氧树脂,即指数s具有>1.5的值。
对于液体环氧树脂,指数s表示小于1的值。s优选表示小于0.2的值。
因此其优选为双酚-a(dgeba)、双酚-f以及双酚-a/f的二缩水甘油醚。所述液体树脂例如可以
还适合作为环氧树脂a的是所谓的环氧线性酚醛清漆。其特别具有下式:
特别地,其在此为苯酚-环氧化物-线性酚醛清漆或甲酚-环氧化物-线性酚醛清漆(r2=ch2)。
所述环氧树脂可以商标名epn或ecn以及
环氧树脂a优选表示式(ii)的液体环氧树脂。
在一个特别优选的实施方案中,热固化环氧树脂组合物不仅包含至少一种式(ii)的液体环氧树脂(其中s<1,特别是小于0.2),而且包含至少一种式(ii)的固体环氧树脂(其中s>1.5,特别是2至12)。
优选地,环氧树脂a的份额为10-60重量%,特别是30-50重量%,以环氧树脂组合物的总重量计。
还有利的是,50-100重量%,特别是80-100重量%的环氧树脂a为上述液体环氧树脂。
还有利的是,0-30重量%,特别是0-20重量%,特别优选5-15重量%的环氧树脂a为上述固体环氧树脂。
根据本发明的组合物还包含双氰胺作为固化剂b。
优选地,双氰胺b具有0.5-50μm、3-15μm、3-12μm,特别是3-10μm,优选4-8μm的粒径d98。
优选地,平均粒径d50为0.5-50μm,特别是1-15μm,优选1-7μm,特别优选2.1-5μm。
出人意料地发现,这在根据本发明的组合物中造成粘合的改进以及冲击剥离强度的改进。这例如在表3中通过更好的拉伸剪切强度和更好的i-剥离值得以证实。还出人意料的是,在根据本发明的组合物中,在2.1-5μm的平均粒径d50下发现特别长的储存稳定性,这通过在50℃下储存一周之后粘度略微升高得以证实。
术语“平均粒径”在此表示累积体积分布曲线的d50-值,其中50体积%的颗粒具有小于所述值的直径。平均粒径或d50-值在本发明中通过激光衍射确定。
在本文中,例如d10、d50、d90和d98表示这样的直径:10体积%、50体积%(“平均粒径”)、90体积%或98体积%的颗粒具有通过激光衍射确定的比其更小的直径。
根据本发明的组合物还包含至少一种促进剂c,其中所述促进剂c为通过如下物质的反应获得的化合物:
-1重量份的胺/环氧-加合物(a),和
-0.1至0.8,优选0.2至0.6重量份的化合物(b),所述化合物(b)选自酚树脂和多元酚化合物;
其中胺/环氧-加合物(a)可通过如下物质和至少一种每个分子具有平均多于一个环氧基团的环氧树脂(3)的反应获得:
-下式的氨基化合物(1)
其中r3和r4彼此独立地各自表示具有1至5个碳原子、特别是1至2个碳原子、特别优选2个碳原子的烷基,并且r5表示具有1至5个碳原子、特别是3个碳原子的亚烷基;或
-优选氨基化合物混合物,所述氨基化合物混合物以70/30至99/1、特别是80/20至97/3的(1)与(2)的重量比包含氨基化合物(1)和1-氨基-4-乙基哌嗪(2);
其中二者的比例使得对于氨基化合物(1)或(1)+(2)中的每当量氨基,环氧树脂(3)中的环氧基团的量为0.8至2.5,特别是1.0至1.6当量。
氨基化合物(1)优选选自二甲基氨基丙基胺、二乙基氨基丙基胺、二丙基氨基丙基胺、二丁基氨基丙基胺、二甲基氨基乙基胺、二乙基氨基乙基胺、二丙基氨基乙基胺和二丁基氨基乙基胺。
特别优选为h2nch2ch2ch2n(c2h5)2(3-(二乙基氨基)丙基胺)或h2nch2ch2ch2n(ch3)2(3-(二甲基氨基)-1-丙基胺),最优选为h2nch2ch2ch2n(c2h5)2(3-(二乙基氨基)丙基胺)。
促进剂c的每分子具有平均多于一个环氧基团的至少一种环氧树脂(3)优选为如上所述的式(ii)的环氧树脂。取代基r'和r”优选为h或ch3,特别是ch3。指数s优选表示≤2,特别是≤1.5,特别是≤1的值。s优选表示小于0.2的值。
化合物(b)选自酚树脂和多元酚化合物。优选的酚树脂和多元酚化合物的示例为由酚和醛制得的酚醛树脂,例如苯酚/甲醛树脂、甲酚/甲醛树脂、双酚a(bpa)/甲醛树脂、双酚f(bpf)/甲醛树脂、烷基苯酚/甲醛树脂及其混合物,特别优选苯酚酚醛树脂和甲酚酚醛树脂以及多元酚化合物例如双酚a、双酚f和间苯二酚。
所述促进剂c例如描述于美国专利no.4,689,390中。
还有利的是,促进剂c具有2-80μm,特别是3-30μm,优选3-16μm,优选3-10μm,特别优选4-8μm的粒径d90。
优选地,平均粒径d50为0.5-50μm,特别是1-15μm,优选1-5μm,特别优选1-3μm。
出人意料地发现,这在根据本发明的组合物中造成机械性能的改进以及抗冲击性的改进。这例如在表2中通过更高的拉伸强度值、断裂伸长值和弹性模量值以及更高的冲击剥离值得以证实。还出人意料地发现,这在根据本发明的组合物中造成储存稳定性的改进。
还有利的是,以环氧树脂组合物的总重量计,根据本发明的组合物具有小于2重量%,特别是小于1重量%,优选小于0.5重量%,特别优选小于0.3重量%,最优选小于0.1重量%的选自如下列表的促进剂:脲、封装叔胺、封端叔胺、潜在咪唑和胺-三氟化硼复合物。
以克/摩尔环氧树脂a的环氧基团计,双氰胺b的比例为3-40g/mol环氧基团,并且以克/摩尔环氧树脂a的环氧基团计,促进剂c的比例为5-35g/mol环氧基团。
优选地,以克/摩尔环氧树脂a的环氧基团计,双氰胺b的比例为3-27,优选3-25g/mol环氧基团。出人意料地发现,这大大降低了在固化时组合物中形成气泡的可能性,气泡的形成会造成粘附值和机械性能的降低。
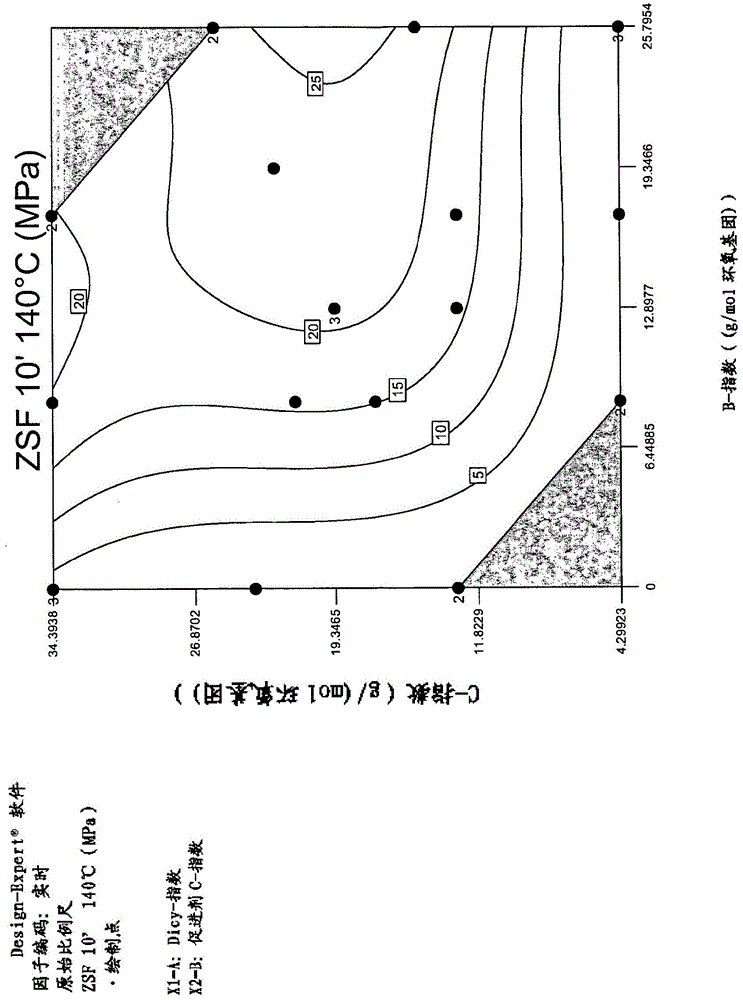
优选地,以克/摩尔环氧树脂a的环氧基团计,双氰胺b的比例为6-30、8-25、12-25、14-25、17-25,优选19-25g/mol环氧基团。出人意料地发现,由此获得高粘附值和特别出人意料的高储存稳定性值。这例如在图1和2中得以证实。
优选地,以克/摩尔环氧树脂a的环氧基团计,促进剂c的比例为10-26、12-25、15-22g/mol环氧基团。
出人意料地发现,由此获得高粘附值。这例如在图1中得以证实。
优选地,以克/摩尔环氧树脂a的环氧基团计,促进剂c的比例为5-26、5-25、5-22、5-20、5-18g/mol环氧基团。
出人意料地发现,由此获得高储存稳定性。这例如在图2中得以证实。
特别优选地,以克/摩尔环氧树脂a的环氧基团计,双氰胺b的比例为6-30、8-30、8-25、12-25、14-25、14-22g/mol环氧基团,并且以克/摩尔环氧树脂a的环氧基团计,促进剂c的比例为10-26、10-25、12-25、12-22、15-20g/mol环氧基团。
这特别有助于良好的粘附和同时长的储存稳定性。
还有利的是,两个比例之和(以克/摩尔环氧树脂a的环氧基团计的双氰胺b的比例)+(以克/摩尔环氧树脂a的环氧基团计的促进剂c的比例)为20-40g/molg/mol环氧基团,优选55-35g/molg/mol环氧基团。当增韧剂d为封端聚氨酯聚合物d1时,这是特别有利的。其优点在于由此获得高粘附值(zsf)。这例如在表4中得以证实。
还有利的是,两个比例之比(以克/摩尔环氧树脂a的环氧基团计的双氰胺b的比例)/(以克/摩尔环氧树脂a的环氧基团计的促进剂c的比例)为0.2-3,特别是0.3-2.5,特别优选0.5-1.5。其优点在于由此获得高粘附值。
特别有利的是,增韧剂d为封端聚氨酯聚合物d1或液体橡胶d2,特别是封端聚氨酯聚合物d1。这例如在表4中得以证实。
热固化环氧树脂组合物任选包含至少一种增韧剂d。所述增韧剂d可以为固体或液体。
特别地,增韧剂d选自封端聚氨酯聚合物d1、液体橡胶d2和核-壳聚合物d3。
如果增韧剂d为封端聚氨酯聚合物d1或核-壳聚合物d3,则这是有利的,因为由此获得高粘附值,特别是在封端聚氨酯聚合物d1的情况下。这例如在表4中得以证实。
还有利的是,增韧剂d为液体橡胶d2或核-壳聚合物d3。其优点在于,由此获得高储存稳定性值,特别是在核-壳聚合物d3的情况下。这例如在表4中得以证实。
还有利的是,增韧剂d为封端聚氨酯聚合物d1或核-壳聚合物d3。其优点在于,由此获得高冲击剥离强度值,特别是在封端聚氨酯聚合物d1的情况下。这例如在表4中得以证实。
在一个实施方案中,增韧剂d为封端聚氨酯聚合物d1,优选为式(i)的封端聚氨酯聚合物。
在此r1表示被异氰酸酯基团封闭的线性或支化的聚氨酯预聚物在除去末端异氰酸酯基团之后的p价-基团,并且p表示2至8的值。
此外,r2彼此独立地表示选自如下基团的取代基
在此r5、r6、r7和r8分别各自独立地表示烷基或环烷基或芳烷基或芳基烷基,或者r5连同r6或r7连同r8形成任选取代的4元环至7元环的一部分。
此外,r9'和r10分别各自独立地表示烷基或芳烷基或芳基烷基或烷氧基或芳氧基或芳烷氧基,并且r11表示烷基。
r12、r13和r14分别各自独立地表示具有2至5个c原子的任选具有双键或被取代的亚烷基,或亚苯基或氢化亚苯基。
r15、r16和r17分别各自独立地表示h或烷基或芳基或芳烷基,并且r18表示芳烷基或任选具有芳族羟基的取代或未取代的单核或多核芳族基团。
最后,r4表示包含伯羟基或仲羟基的脂族、脂环族、芳族或芳脂族环氧化物在除去羟基和环氧基团之后的基团,并且m表示1、2或3的值。
作为r18,一方面特别考虑酚或多酚,特别是双酚在除去羟基之后的基团。所述酚和双酚的优选示例特别是苯酚、甲酚、间苯二酚、焦儿茶酚、腰果酚(3-十五烯基酚(得自腰果壳油))、壬基酚、与苯乙烯或二环戊二烯反应的酚、双酚-a、双酚-f和2,2'-二烯丙基-双酚-a。作为r18,另一方面特别考虑羟基苯甲醇和苯甲醇在除去羟基之后的基团。
当r5、r6、r7、r8、r9、r9’、r10、r11、r15、r16或r17表示烷基时,其特别是线性或支化的c1-c20-烷基。
当r5、r6、r7、r8、r9、r9'、r10、r15、r16、r17或r18表示芳烷基时,所述基团特别是通过亚甲基结合的芳族基团,特别是苯甲基。
当r5,r6,r7,r8,r9,r9'或r10表示烷基芳基时,其特别是通过亚苯基结合的c1-至c20-烷基,例如甲苯基或二甲苯基。
基团r2优选为下式的取代基
作为式
作为式
基团y在此表示具有1至20个c原子,特别是1至15个c原子的饱和芳族或烯属不饱和烃基。特别优选作为y的是烯丙基、甲基、壬基、十二烷基、苯基、烷基醚、羧酸酯或具有1至3个双键的不饱和c15-烷基。
r2最优选表示
由异氰酸酯基团封端的线性或支化聚氨酯预聚物与一种或多种异氰酸酯反应性化合物r2h制备式(i)的封端聚氨酯预聚物。如果使用多种所述异氰酸酯反应性化合物,则反应可以依次进行或者用这些化合物的混合物进行。
优选进行所述反应使得一种或多种异氰酸酯反应性化合物r2h以化学计量比或化学计量过量使用,从而保证所有nco-基团都反应。
具有异氰酸酯端基的聚氨酯预聚物(r1即基于此)可以由至少一种二异氰酸酯或三异氰酸酯以及由具有末端氨基、硫醇基或羟基的聚合物qpm和/或由任选取代的多酚qpp制得。
合适的二异氰酸酯为脂族、脂环族、芳族或芳脂族的二异氰酸酯,特别是市售产品例如亚甲基二苯基二异氰酸酯(mdi)、六亚甲基二异氰酸酯(hdi)、甲苯二异氰酸酯(tdi)、联甲苯胺二异氰酸酯(todi)、异佛尔酮二异氰酸酯(ipdi)、三甲基六亚甲基二异氰酸酯(tmdi)、2,5-或2,6-双-(异氰酸基甲基)-双环[2.2.1]庚烷、1,5-萘二异氰酸酯(ndi)、二环己基甲基二异氰酸酯(h12mdi)、对-亚苯基二异氰酸酯(ppdi)、间-四甲基苯二亚甲基二异氰酸酯(tmxdi)等及其二聚物。优选的为hdi、ipdi、mdi或tdi。
合适的三异氰酸酯为脂族、脂环族、芳族或芳脂族二异氰酸酯的三聚物或缩二脲,特别是上一段落所述的二异氰酸酯的异氰脲酸酯和缩二脲。当然还可以使用二异氰酸酯或三异氰酸酯的合适的混合物。
特别适合作为具有末端氨基、硫醇基或羟基的聚合物qpm的是具有两个或三个末端氨基、巯基或羟基的聚合物qpm。
聚合物qpm有利地具有300-6000,特别是600-4000,优选700-2200g/当量nco-反应性基团的当量。
适合作为聚合物qpm的是多元醇,例如如下市售多元醇或其任何混合物:
-聚氧化亚烷基多元醇(也被称为聚醚多元醇),其为环氧乙烷、1,2-环氧丙烷、1,2-或2,3-环氧丁烷、四氢呋喃或其混合物的任选借助于具有两个或三个活性h-原子的起始分子聚合的聚合产物,所述起始分子例如为水或具有两个或三个oh-基团的化合物。可以使用例如借助于所谓的双金属氰络合物催化剂(简称dmc-催化剂)制备的具有低不饱和度(根据astmd-2849-69测量并且用毫当量不饱和/克多元醇(meq/g)表示)的聚氧化亚烷基多元醇,也可以使用例如借助于阴离子型催化剂(例如naoh、koh或碱金属醇盐)制备的具有更高不饱和度的聚氧化亚烷基多元醇。特别合适的是不饱和度低于0.02meq/g并且平均分子量为1000-30000道尔顿的聚氧化亚丙基二醇和三醇,平均分子量为400-8000道尔顿的聚氧化亚丁基二醇和三醇、聚氧化亚丙基二醇和三醇,以及所谓的“eo-封端”(环氧乙烷封端)的聚氧化亚丙基二醇或三醇。后者特别为聚氧化亚丙基聚氧化亚乙基多元醇,其例如这样获得:在聚丙氧基化结束之后用环氧乙烷烷氧基化纯的聚氧化亚丙基多元醇,因此使其具有伯羟基。
-羟基封端的聚丁二烯多元醇,例如通过1,3-丁二烯和烯丙基醇的聚合或者通过聚丁二烯的氧化制得的那些,及其氢化产物;
-苯乙烯-丙烯腈接枝的聚醚多元醇,例如由basf以名称
-多羟基封端的丙烯腈/丁二烯-共聚物,例如可以由羧基封端的丙烯腈/丁二烯-共聚物(以名称
-例如由二元醇至三元醇(例如1,2-乙二醇、二乙二醇、1,2-丙二醇、二丙二醇、1,4-丁二醇、1,5-戊二醇、1,6-己二醇、新戊二醇、甘油、1,1,1-三羟甲基丙烷或上述醇的混合物)与有机二羧酸或其酸酐或酯(例如琥珀酸、戊二酸、己二酸、辛二酸、癸二酸、十二烷二羧酸、马来酸、富马酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸和六氢邻苯二甲酸或上述酸的混合物)制得的聚酯多元醇,以及得自内酯例如ε-己内酯的聚酯多元醇;
-聚碳酸酯多元醇,例如通过例如上述为了构建聚酯多元醇所使用的醇与碳酸二烷基酯、碳酸二芳基酯或碳酰氯的反应获得的那些。
有利地,聚合物qpm为oh-当量为300至6000g/oh-当量,特别是600至4000g/oh-当量,优选700-2200g/oh-当量的双官能或更多官能的多元醇。还有利的是,多元醇选自聚乙二醇、聚丙二醇、聚乙二醇-聚丙二醇-嵌段共聚物、聚丁二醇、羟基封端的聚丁二烯、羟基封端的丁二烯/丙烯腈-共聚物、羟基封端的合成橡胶、其氢化产物和所述多元醇的混合物。
此外作为聚合物qpm,还可以使用双官能或更多官能的氨基封端的聚乙烯醚,聚丙烯醚,例如以名称
对于某些用途,适合作为聚合物qpm的特别是具有羟基的聚丁二烯或聚异戊二烯或其部分或完全氢化的反应产物。
还有可能的是,聚合物qpm还可以是链延长的,正如以本领域技术人员已知的方式和方法通过多胺、多元醇和多异氰酸酯(特别是二胺、二醇和二异氰酸酯)的反应所进行的。
例如二异氰酸酯和二醇如下所示根据所选择的化学计量比形成式(vi)或(vii)的物质
基团y1和y2表示二价有机基团并且指数u和v根据化学计量比从1至通常5而变化。
式(vi)或(vii)的这些物质然后可以重新继续反应。因此可以例如由式(vi)的物质和具有二价有机基团y3的二醇形成下式的链延长的聚氨酯预聚物:
可以由式(vii)的物质和具有二价有机基团y4的二异氰酸酯形成下式的链延长的聚氨酯预聚物:
指数x和y根据化学计量比从1至通常5而变化并且特别是1或2。
此外,式(vi)的物质也可以与式(vii)的物质反应,从而形成具有nco-基团的链延长的聚氨酯预聚物。
为了链延长,特别优选二醇和/或二胺和二异氰酸酯。本领域技术人员当然清楚,为了链延长也可以使用更高官能的多元醇(例如三羟甲基丙烷或季戊四醇)或更高官能的多异氰酸酯(例如二异氰酸酯的异氰脲酸酯)。
一般对于聚氨酯预聚物并且特别对于链延长的聚氨酯预聚物而言,有利地需要注意预聚物不具有过高粘度,特别是当将更高官能的化合物用于链延长时,因为过高粘度可能使其难以反应形成式(i)的聚氨酯预聚物或者使得粘合剂的施用变困难。
优选作为聚合物qpm的是选自如下的平均分子量在600和6000道尔顿之间的多元醇:聚乙二醇、聚丙二醇、聚乙二醇-聚丙二醇-嵌段聚合物、聚丁二醇、羟基封端的聚丁二烯、羟基封端的丁二烯-丙烯腈-共聚物及其混合物。
特别优选作为聚合物qpm的是具有c2-c6-亚烷基或具有混合的c2-c6-亚烷基的用氨基、巯基或优选羟基封端的α,ω-二羟基聚亚烷基二醇。特别优选的是聚丙二醇或聚丁二醇。还特别优选的是羟基封端的聚氧化丁烯。
特别适合作为多酚qpp的是双酚、三酚和四酚。其不仅被理解为纯酚,而且任选还被理解为取代的酚。取代基的种类可以非常多样。特别地,其被理解为直接位于与酚oh-基团结合的芳族核上的取代基。此外,酚不仅被理解为单核芳族化物,还被理解为多核或稠合的芳族化物或杂芳族化物,其具有直接位于芳族化物或杂芳族化物上的酚oh-基团。
这些取代基的种类和位置特别影响为了形成聚氨酯预聚物所需的与异氰酸酯的反应。
特别合适的是双酚和三酚。例如适合作为双酚或三酚的是1,4-二羟基苯、1,3-二羟基苯、1,2-二羟基苯、1,3-二羟基甲苯、3,5-二羟基苯甲酸酯、2,2-双(4-羟基苯基)丙烷(=双酚-a)、双(4-羟基苯基)甲烷(=双酚-f)、双(4-羟基苯基)砜(=双酚-s)、萘酚间苯二酚、二羟基萘、二羟基蒽醌、二羟基-联苯、3,3-双(对-羟基苯基)苯酞、5,5-双(4-羟基苯基)六氢-4,7-亚甲基茚满、酚酞、荧光素、4,4'-[双-(羟基苯基)-1,3-亚苯基双-(1-甲基-亚乙基)](=双酚-m)、4,4'-[双-(羟基苯基)-1,4-亚苯基双-(1-甲基-亚乙基)](=双酚-p)、2.2'-二烯丙基-双酚-a、通过甲酚或甲酚与二异亚丙基苯的反应制得的双酚和二甲酚、间苯三酚、没食子酸酯、oh-官能度为2.0至3.5的酚醛清漆或甲酚酚醛清漆以及上述化合物的所有异构体。
在第一个实施方案中,聚氨酯预聚物由至少一种二异氰酸酯或三异氰酸酯以及具有末端氨基、硫醇基或羟基的聚合物qpm制得。聚氨酯预聚物的制备以聚氨酯领域技术人员已知的方式和方法进行,特别地,其中二异氰酸酯或三异氰酸酯相对于聚合物qpm的氨基、硫醇基或羟基以化学计量过量使用。
在第二个实施方案中,聚氨酯预聚物由至少一种二异氰酸酯或三异氰酸酯以及任选取代的多酚qpp制得。聚氨酯预聚物的制备以聚氨酯领域技术人员已知的方式和方法进行,特别地,其中二异氰酸酯或三异氰酸酯相对于多酚qpp的酚基以化学计量过量使用。
在第三个实施方案中,聚氨酯预聚物由至少一种二异氰酸酯或三异氰酸酯以及具有末端氨基、硫醇基或羟基的聚合物qpm以及任选取代的多酚qpp制得。为了由至少一种二异氰酸酯或三异氰酸酯以及具有末端氨基、硫醇基或羟基的聚合物qpm和/或由任选取代的多酚qpp制备聚氨酯预聚物,可以采用多种不同的途径。
特别优选的是第一个实施方案。
具有异氰酸酯基团的聚氨酯预聚物优选具有弹性特征。其优选具有小于0℃的玻璃化转变温度tg。
在另一个实施方案中,所述增韧剂d为液体橡胶d2,其特别是被羧基或(甲基)丙烯酸酯基团或环氧基团封端的丙烯腈/丁二烯-共聚物或其衍生物。这种液体橡胶例如以emeraldperformancematerialsllc的名称hyprotm(之前为
本领域技术人员清楚的是,当然也可以使用液体橡胶的混合物,特别是羧基封端或环氧化物封端的丙烯腈/丁二烯-共聚物或其衍生物与环氧化物封端的聚氨酯预聚物的混合物。
在另一个实施方案中,所述增韧剂d为核/壳聚合物d3。核-壳聚合物由弹性核聚合物和刚性壳聚合物组成。特别合适的核/壳聚合物由被刚性热塑性聚合物的壳包围的弹性丙烯酸酯聚合物或丁二烯聚合物的核组成。所述核-壳结构通过嵌段共聚物的分离自发形成,或者通过乳液聚合或悬浮聚合形式的聚合进程与随后的接枝而形成。优选的核-壳聚合物为所谓的mbs聚合物,其以atofina的商标名clearstrengthtm,rohm的商标名paraloidtm和zeon的商标名f-351tm市售获得。
特别优选的是可能以悬浮体形式存在的核-壳聚合物颗粒。其示例为wacker的具有聚硅氧烷核和丙烯酸酯壳的genioperlm23a,eliokem制备的nep系列的辐射交联的橡胶颗粒,或lanxess的nanoprene或rohmundhaas的paraloidexl或kaneka的kaneacemx-120。
核-壳聚合物的其它相当的示例以德国evonikhansegmbh的名称albidur提供。
同样适合的是环氧化物基质中的纳米级硅酸盐,例如以德国evonikhansegmbh的商标名nanopox销售的那些。
份额优选为:
增韧剂d110-60重量%,特别是20-30重量%;
增韧剂d210-30重量%,特别是20-30重量%;
增韧剂d310-30重量%,特别是20-30重量%;
以环氧树脂组合物的总重量计。
在另一个优选的实施方案中,组合物还包含至少一种填料f。其优选为云母、滑石、高岭土、钙硅石、长石、正长岩、绿泥石、斑脱土、蒙脱土、碳酸钙(沉淀或研磨)、白云石、石英、二氧化硅(热解或沉淀)、方晶石、氧化钙、氢氧化铝、氧化镁、陶瓷空心球、玻璃空心球、有机空心球、玻璃球、着色颜料。特别优选的是选自碳酸钙、氧化钙和热解二氧化硅的填料。
有利地,所有填料f的总份额为5-40重量%,优选10-30重量%,以环氧树脂组合物的总重量计。
在另一个优选的实施方案中,组合物可以包含物理发泡剂或化学发泡剂,如例如以akzonobel公司的商标名expancel或chemtura公司的celogen或lehmann&voss的商标名
在另一个优选的实施方案中,组合物还包含至少一种带有环氧基团的反应性稀释剂g。所述反应性稀释剂是本领域技术人员已知的。带有环氧基团的反应性稀释剂的优选示例为:
-单官能饱和或不饱和、支化或非支化、环状或开链的c4-c30醇的缩水甘油醚,例如丁醇缩水甘油醚、己醇缩水甘油醚、2-乙基己醇缩水甘油醚、烯丙基缩水甘油醚、四氢糠基缩水甘油醚和糠基缩水甘油醚、三甲氧基甲硅烷基缩水甘油醚等。
-双官能饱和或不饱和、支化或非支化、环状或开链的c2-c30醇的缩水甘油醚,例如乙二醇缩水甘油醚、丁二醇缩水甘油醚、己二醇缩水甘油醚、辛二醇缩水甘油醚、环己烷二甲醇二缩水甘油醚、新戊二醇二缩水甘油醚等。
-三官能或多官能的饱和或不饱和、支化或非支化、环状或开链的醇(例如环氧化蓖麻油、环氧化三羟甲基丙烷、环氧化季戊四醇)的缩水甘油醚,或脂族多元醇(例如山梨醇、甘油或三羟甲基丙烷)的聚缩水甘油醚等。
-酚化合物和苯胺化合物的缩水甘油醚,例如苯基缩水甘油醚、甲苯基缩水甘油醚、对叔丁基苯基缩水甘油醚、壬基酚缩水甘油醚、3-正十五烯基-缩水甘油醚(得自腰果壳油)、n,n-二缩水甘油基苯胺等。
-环氧化的胺,例如n,n-二缩水甘油基环己胺等。
-环氧化单羧酸或二羧酸,例如新癸酸-缩水甘油酯、甲基丙烯酸缩水甘油酯、苯甲酸缩水甘油酯、邻苯二甲酸二缩水甘油酯、四氢和六氢邻苯二甲酸二缩水甘油酯、二聚脂肪酸的二缩水甘油酯等。
-环氧化的二官能或三官能、低分子量至高分子量的聚醚多元醇,例如聚乙二醇-二缩水甘油醚、聚丙二醇-二缩水甘油醚等。
特别优选的是己二醇二缩水甘油醚、甲苯基缩水甘油醚、对-叔丁基苯基缩水甘油醚、聚丙二醇二缩水甘油醚和聚乙二醇二缩水甘油醚。
有利地,带有环氧基团的反应性稀释剂g的总份额为0.1-15重量%,优选0.1-5重量%,特别优选0.1-2重量%,特别优选0.2-1重量%,以环氧树脂组合物的总重量计。
组合物可以包含其它成分,特别是催化剂,稳定剂,特别是热稳定剂和/或光稳定剂,触变剂,增塑剂,溶剂,矿物填料或有机填料,发泡剂,着色剂和颜料,防腐蚀剂,表面活性剂,消泡剂和增粘剂。
适合作为增塑剂的特别是苯酚烷基磺酸酯或苯磺酸-n-丁酰胺,例如以
适合作为稳定剂的特别是任选取代的苯酚,例如bht或
特别优选的单组份环氧树脂组合物包含:
-以环氧树脂组合物的总重量计,10-60重量%、特别是30-50重量%的每分子具有平均多于一个环氧基团的环氧树脂a;优选地,50-100重量%、特别是80-100重量%的环氧树脂a为液体环氧树脂并且0-30重量%、特别是0-20重量%、特别优选5-15重量%的环氧树脂a为固体环氧树脂;
-双氰胺b;
-至少一种促进剂c,其中促进剂c为每分子具有平均多于一个环氧基团的至少一种环氧树脂ah与具有至少一个伯氨基和至少一个叔氨基的至少一种脂族多胺pa的反应产物;
-至少一种增韧剂d,选自封端的聚氨酯聚合物d1、液体橡胶d2和核-壳聚合物d3,份额优选为:
增韧剂d110-60重量%,特别是20-30重量%;
增韧剂d210-30重量%,特别是20-30重量%;
增韧剂d310-30重量%,特别是20-30重量%;
以环氧树脂组合物的总重量计;
-以环氧树脂组合物的总重量计,优选5-40重量%、优选10-30重量%的填料f,选自碳酸钙、氧化钙和热解二氧化硅;
-以环氧树脂组合物的总重量计,优选0.1-15重量%、优选0.1-5重量%、特别优选0.1-2重量%、特别优选0.2-1重量%的带有环氧基团的反应性稀释剂g;
-其中,以克/摩尔环氧树脂a的环氧基团计,双氰胺b的比例为6-30、8-30、8-25、12-25、14-25、14-22g/mol环氧基团,并且以克/摩尔环氧树脂a的环氧基团计,促进剂c的比例为10-26、10-25、12-25、12-22、15-20g/mol环氧基团。
还有利的是,以环氧树脂组合物的总重量计,优选的单组份环氧树脂组合物的大于80重量%,优选大于90重量%,特别是大于95重量%,特别优选大于98重量%,最优选大于99重量%由上述成分组成。
特别优选的组合物的实施例为表3中的ex.6。
有利的是,根据本发明的环氧树脂组合物在25℃下具有500-3000pa*s,特别是1000-2500pa*s,优选1000-2000pa*s的粘度。其优点在于由此保证良好的可施用性。
还有利的是,根据本发明的环氧树脂组合物在制备后一天在50℃下储存1周之后在25℃的测量温度下测得的粘度的升高量小于200%,小于150%,小于120%,小于100%,小于50%。
已经发现,所述热固化环氧树脂组合物特别适合用作单组份热固化粘合剂,特别是在车辆构造中用作热固化单组份车身粘合剂。这种单组份粘合剂具有广泛的应用可能性。特别地,由此可实现在较高温和低温下都具有高抗冲性的热固化单组份粘合剂。这种粘合剂对于粘合热稳定性材料来说是需要的。热稳定性材料被理解为在100-220℃,优选120-200℃的固化温度下至少在固化时间内形状稳定的材料。其特别为金属和塑料例如abs、聚酰胺、聚苯醚,复合材料例如smc、不饱和聚酯gfk、环氧化物复合材料或丙烯酸酯复合材料。优选的是至少一种材料为金属的应用。特别优选的应用是粘合特别是汽车工业中的车身中的相同或不同的金属。优选的金属主要是钢,特别是电镀锌、热镀锌、涂油钢,涂覆博纳锌的钢,和随后磷化的钢,以及铝,特别是在汽车制造中通常出现的变体形式。
使用基于根据本发明的热固化组合物的粘合剂能够实现不仅在高固化温度下而且在低固化温度下的高冲击强度的希望的组合。
特别地,这种粘合剂首先在10℃和80℃之间,特别是10℃和60℃之间的温度下与待粘合材料接触,然后在通常130-220℃,优选130-180℃,特别优选130-150℃的温度下固化。
本发明的另一个方面涉及用于粘合热稳定性基材的方法,包括如下步骤:
i)将如上详细描述的热固化环氧树脂组合物施用至热稳定性基材s1、特别是金属的表面;
ii)使施用的热固化环氧树脂组合物与另一个热稳定性基材s2、特别是金属的表面接触;
iii)将组合物加热至100-220℃,特别是120-200℃,优选130和150℃之间,特别优选130和140℃之间的温度。
基材s2在此由与基材s1相同或不同的材料组成。
基材s1和/或s2特别是上述金属和塑料。
优选地,在步骤iii)中将组合物加热至100-220℃,特别是120-200℃,优选130和150℃之间,特别优选130和140℃之间的温度,使组合物在上述温度下保持10min-6h、10min-2h、10min-60min、10min-30min、10min-20min,特别优选10min-15min。
通过这种用于粘合热稳定性材料的方法获得经粘合制品。所述制品优选为车辆或车辆的部件。
本发明的另一个方面因此涉及由上述方法获得的经粘合制品。当然,除了热固化粘合剂之外,通过根据本发明的组合物还可以实现密封料。此外,根据本发明的组合物不仅适合车辆构造而且适合其它应用领域。特别要提及的是在运输工具(例如船、货车、大客车或轨道车辆)或日用品制造(例如洗衣机)中的相关应用。
经由根据本发明的组合物粘合的材料可以在通常120℃和-40℃之间,优选100℃和-40℃之间,特别是80℃和-40℃之间的温度下使用。
根据本发明的热固化环氧树脂组合物的特别优选的用途是其在车辆构造中作为热固化单组份车身粘合剂或作为硬化料或在结构构件和增强元件的空腔中作为用于增强的可发泡热固化组合物的用途。
本发明的另一个方面涉及经固化的环氧树脂组合物,例如通过加热如上具体描述的热固化环氧树脂组合物获得的经固化环氧树脂组合物。加热通常在炉中在100-220℃,优选130和150℃之间,特别优选130和140℃之间的温度下进行,优选在上述温度下进行10min-6h、10min-2h、10min-60min、10min-30min、10min-20min,特别优选10min-15min。
已经发现,在固化温度下,特别在130和150℃之间的固化温度下,促进剂c(如上文作为热固化环氧树脂组合物的成分已经详细描述过的)适合作为热固化环氧树脂组合物的促进剂。
出人意料地发现,用于热固化环氧树脂组合物的其它促进剂不满足低温下促进和同时有储存稳定性的要求。因此例如在表1中所示,例如当使用封装的叔胺(lc-100,a&ccatalystsinc)或环氧树脂与咪唑的反应产物(a&ccatalystsinc.)时,相应环氧树脂组合物的储存稳定性剧烈变差。此外,相比于根据本发明的促进剂,130℃的固化温度下的粘附更差。
另一方面,使用bf3-胺-加合物(
还可以确定的是,通过在热固化环氧树脂组合物中加入促进剂c,相比于不使用促进剂c的情况,其可以在明显更低的温度下固化。
出人意料地发现,使用增韧剂d、特别是增韧剂d1改进了粘附。因此在表5中可看出,不具有增韧剂的热固化环氧树脂组合物具有较差的粘附,这例如通过拉伸剪切强度以及t-剥离的值所看出。还出人意料地发现,所述环氧树脂组合物具有降低的储存稳定性。
实施例
下文列出一些实施例,所述实施例进一步说明本发明但是不应当以任何方式限制本发明的范围。
增韧剂(“d-1”)的制备
将150gpoly-thf2000(oh-数57mg/gkoh)和150liquiflexh(oh-数46mg/gkoh)在105℃下真空干燥30分钟。在温度已降低至90℃之后,加入61.5gipdi和0.14g二月桂酸二丁基锡。在90℃下真空进行反应直至2.0h后nco-含量恒定于3.10%(计算的nco-含量:3.15%)。然后加入96.1g腰果酚作为封端剂。在105℃下真空继续搅拌直至不再检测到游离nco。产物作为增韧剂d-1使用。
使用的原料
组合物的制备
根据表1-7中的数据制备参考组合物ref.1-ref.11以及根据本发明的组合物ex.1-ex.27。表1-5中的用量数据以克计,表6中的用量数据以重量份计。
对于组合物(其值显示于图1和2以及表7中)的制备,分别使用根据表6的组合物,其中仅改变b或c的以重量份计的量从而获得x-轴中所示的b-指数或y-轴中所示的c-指数。a-液体树脂、反应性稀释剂、a-固体树脂、d1以及填料混合物的重量份都不改变。在表6所示的实施例中,例如总共获得106.25重量份。
图1中显示的测量值以mpa给出,标记“20”的线表示20mpa的值。图2中显示的测量值对应于储存后相比于储存前的值的粘度升高。标记“0.5”的线表示粘度升高50%,标记“1.5”的线表示粘度升高150%。伴随数字“2”或“3”的测量点为两次或三次确定的测量值。图1和图2中显示的测量值还示于表7中。
例如通过表7可见,具有比根据本发明的更低的b-指数或c-指数的参考组合物ref.6-ref.11具有不足的拉伸剪切强度值。此外在参考组合物ref.8中,储存稳定性相比于根据本发明的组合物明显降低。
b-指数、c-指数、总和(b+c-指数)和比例(b/c-指数)的计算
环氧树脂根据其环氧含量进行划分,所述环氧含量也被称为环氧数(即ep-数)。在本文中,环氧数表示100克合成树脂中存在的环氧基团的摩尔数。因此例如在表中,作为环氧数,通过“ep-数”将例如0.54mol环氧基团/100g树脂的环氧数简化成“0.54”的数值。
例如对于组合物ex.6,以克/摩尔环氧树脂a的环氧基团计的双氰胺b的比例(在表和图1和2中称为“b-指数”)以如下方式计算。
(双氰胺的以克计的量)/((a-液体树脂以克计的量*a-液体树脂的环氧数)+(反应性稀释剂的以克计的量*反应性稀释剂的环氧数)+(a-固体树脂以克计的量*a-固体树脂的环氧数))。
具体为
(3.41g)/((38.42g*0.54mol环氧基团/100g树脂)+(0.48g*0.80mol环氧基团/100g树脂)+(3.84g*0.315mol环氧基团/100g树脂))=15.26(g/mol环氧基团)。
例如对于组合物ex.6,以克/摩尔环氧树脂a的环氧基团计的促进剂c的比例(在表和图1和2中称为“c-指数”)以如下方式计算。
(促进剂此处为促进剂c的以克计的量)/((a-液体树脂以克计的量*a-液体树脂的环氧数)+(反应性稀释剂的以克计的量*反应性稀释剂的环氧数)+(a-固体树脂以克计的量*a-固体树脂的环氧数))。
具体为
(3.84g)/((38.42g*0.54mol环氧基团/100g树脂)+(0.48g*0.80mol环氧基团/100g树脂)+(3.84g*0.315mol环氧基团/100g树脂))=17.20(g/mol环氧基团)。
b-指数和c-指数的值之和被称为“总和(b+c-指数)”,例如对于组合物ex.6以如下方式计算:15.26+17.20(g/mol环氧基团)=32.46(g/mol环氧基团)
b-指数的值除以c-指数的值的比例在表中被称为“比例(b/c-指数)”,例如对于组合物ex.6以如下方式计算:15.26(g/mol环氧基团)/17.20(g/mol环氧基团)=0.89。
测试方法:
拉伸强度,断裂伸长和弹性模量(dineniso527)
在两个聚四氟乙烯纸之间将粘合剂样品挤压成2mm的层厚度。在175℃下固化35分钟之后除去聚四氟乙烯纸并且根据din-标准状态冲压样品。在标准气候下以2mm/min的拉伸速度测量样品。根据dineniso527确定拉伸强度(zf)、断裂伸长和弹性模量0.05-0.25%。
拉伸剪切强度(zsf)(dinen1465)
使用粘合剂以0.3mm的层厚度使经清洗并且用anticoritpl3802-39s背面涂油的eloh420钢制测试板(厚度1.5mm)在25x10mm的粘合表面上与作为间隔件的玻璃珠粘合,并且在175℃的炉温下固化35分钟,或者在140℃的炉温下固化10分钟,或者在135℃的炉温下固化10分钟,或者在130℃的炉温下固化10分钟。在试样表面(基材表面)分别固定温度探针。当达到精确至1℃的给定温度时分别开始测量固化时间。
在拉伸机上以10mm/min的拉伸速度通过3次确定根据dinen1465来确定拉伸强度。
冲击剥离强度/冲击剥离(i-剥离)(根据iso11343)
用粘合剂和尺寸为90x20x0.8mm的钢dc04+ze制备样品。在此,粘合表面为20x30mm,层厚度为0.3mm,玻璃珠充当间隔件。样品在140℃的炉温下固化10分钟,或者在135℃的炉温下固化10分钟,或者在130℃的炉温下固化10分钟。在试样表面(基材表面)分别固定温度探针。当达到精确至1℃的给定温度时分别开始测量固化时间。
在zwick450冲击摆锤上分别在给定温度(23℃、-30℃)下以三次确定的方式进行冲击剥离强度的测量。给出的冲击剥离强度是根据iso11343从25%至90%的测量曲线下的单位为n/mm的平均力。
角剥离强度(t-剥离)(din53281)
制备130x30mm的dc-04+ze钢制测试板(厚度0.8mm)。用合适的冲压机使测试板在30mm的高度处弯曲(90°)。使用粘合剂以0.2mm的层厚度粘合经清洗并且用anticoritpl3802-39s背面涂油的100x30mm的表面和作为间隔件的玻璃珠,并且在140℃的炉温下固化10分钟,或者在135℃的炉温下固化10分钟,或者在130℃的炉温下固化10分钟。在试样表面(基材表面)分别固定温度探针。当达到精确至1℃的给定温度时分别开始测量固化时间。在拉伸机上以50mm/min的拉伸速度通过3次确定以单位为n/mm的剥离力的形式在1/6至5/6行程的横向路径的范围内确定角剥离强度。
粘合剂的粘度/储存稳定性
制备1天之后在生产商antonpaar的mcr101型流变仪上通过使用板-板几何形状在25℃或50℃的温度下以如下参数以振荡方式进行粘合剂上的粘度测量:5hz,1mm间隙,板-板-距离25mm,1%变形。
为了评估粘合剂的储存稳定性,在给定温度下储存一周给定时间之后重复进行粘度测量,并且确定储存后造成的粘度升高百分比。
表1,cured=经固化,uncured=未固化,n.b.=未确定
表2
表3
表5
表6
表7。
- 还没有人留言评论。精彩留言会获得点赞!