一种金属配合物、中间体、其制备方法及应用与流程
本发明涉及一种金属配合物、中间体、其制备方法及应用。
背景技术:
从二十世纪五十年代发现了ziegler-natta催化剂以来,高活性的mgcl2负载的钛的催化剂显示了很好的催化性能(k.ziegler等,angew.chem.1995,67,424;k.ziegler等,angew.chem.1995,67,541;n.kashiwa等,usp-3642746,1968),目前工业上,这种催化剂已经用于高密度聚乙烯(hdpe),线性低密度聚乙烯(lldpe),间规聚丙烯(i-pp)的聚合物的生产。然而,这种多活性中心的固体催化剂目前还不能很好的通过调节催化剂结构来控制聚合物结构、性能;第四族茂金属催化剂的发现则较好地解决了这个问题,由于具有单活性中心,使人们能够根据需要通过改变催化剂的结构来得到预期结构的聚合物(w.kaminsky等,adv.organomet.chem.1980,18,99;w.kaminsky等,angew.chem.,int.ed.engl.1980,19,390;h.h.brintzinger等,angew.chem.int.ed.engl.1995,34,1143)。除此以外,聚烯烃表面能低、化学惰性,对材料进行粘黏,染色,印刷和共混(添加剂、极性材料)时受到限制,因此需要通过在聚烯烃高分子链上引入少量极性官能团增强聚烯烃的极性,改善聚烯烃材料与极性材料的相容性,同时保持相当的物理机械性能。但是前过渡金属催化剂,包括已经广为应用ziegler-natta催化剂和茂金属催化剂,均很容易被极性单体中的极性基团毒化而失活,使得乙烯与极性单体共聚结果仍不理想,如存在插入效率低、大宗极性烯烃单体难以参与共聚合等问题。
近十几年来,以含n、o、p等配位原子的配体代替环戊二烯与过渡金属配位得到的金属配合物作为烯烃聚合催化剂的研究也蓬勃发展起来,这一类催化剂被统称为“茂后催化剂”,其中代表性的过渡金属配合物有如下几种:
文献中已经报道的后过渡金属配位的“茂后催化剂”由于中心金属的亲氧性较弱,可以实现烯烃与极性单体的共聚,能够生产出性能优异的功能化聚烯烃材料,但活性偏低(例如:a)johnson,l.k.etal.j.am.chem.soc.,1996,118(1),267;b)mecking,s.etal.j.am.chem.soc.,1998,120(5),888;c)chien,j.c.w.etal.polym.int.,2002,51,729;d)connor,e.f.etal.j.polym.sci.:parta:polym.chem,2002,40,2842;e)britovesk,g.j.p.etal.j.chem.soc.,daltontrans.,2002,1159;f)chen,g.etal.j.am.chem.soc.,2003,125(22),6697;g)drent,e.;vandijk,r.;vanginkel,r.;vanoort,b.;pugh,r.i.chem.commun.2002,744;h)nakamura,a.;anselment,t.m.j.;claverie,j.;goodall,b.;jordan,r.f.;mecking,s.;rieger,b.;sen,a.;vanleeuwen,p.w.n.m.;nozaki,k.acc.chem.res.2013,46,1438.)。
近年来,科学家相继报道了一些多核烯烃聚合催化剂,中心金属包括ti、cr、zr等前过渡金属以及ni、pd、co、fe等后过渡金属。这类多核金属催化剂通过多个共价键将多个活性金属中心相连,在催化烯烃聚合过程中表现出一些独特的效应(例如:a)li,l.;metz,m.v.;li,h.;chen,m.-c.;marks,t.j.;liable-sands,l.;rheingold,a.l.j.am.chem.soc.2002,124,12725;b)li,h.;li,l.;marks,t.j.angewchem.,int.ed.2004,43,4937;c)salata,m.r.;marks,t.j.j.am.chem.soc.2008,130,12;d)(a)liu,s.;motta,a.;delferro,m.;marks,t.j.j.am.chem.soc.2013,135,8830.(b)liu,s.;motta,a.;delferro,m.;marks,t.j.j.am.chem.soc.2014,136,10460)。
目前,本领域中在能制备得到高分子量的聚合物的同时,又具有插入效率高、催化活性高等方面优势的聚烯烃催化剂仍有待进一步开发。
技术实现要素:
本发明的目的是提供一种金属配合物、中间体、其制备方法及应用。采用本发明的金属配合物可单独或在助催化剂的作用下,用于催化乙烯的均聚合,催化乙烯均聚合,催化活性高(可达>106g/(molmetal)·h),所制备聚乙烯具有高分子量高(可达mn=105~106g/mol)、分子量分布较窄(pdi=1.0~2.5)的特点;此外,制备的聚乙烯含有烷烃支链(1~20支链/1000碳),所述的烷烃支链包含甲基支链、乙基支链、丙基支链以及大于c4的支链。所述的烷烃支链中甲基支链比例为0.75-0.95,乙基支链含量0.01-0.05,丙基支链含量0.01-0.05,大于c4的支链含量0.01-0.10;聚合物熔融温度tm=70~140℃。
本发明提供了一种如式i所示的金属配合物:
其中,→为配位键;
r1、r2、r3、r4、r5和r6各自独立地为:氢、-cf3、r1-1取代或未取代的c1~c10的烷基(所述的r1-1的个数可为一个或多个[例如2个或3个],当存在多个r1-1时,任意两个r1-1相同或不同;所述的“c1~c10的烷基”例如c1~c6的烷基,又例如甲基、乙基、丙基、丁基或己基;)、r1-2取代或未取代的c6~c14的芳基(所述的r1-2的个数可为一个或多个[例如2个或3个],当存在多个r1-2时,任意两个r1-2相同或不同;所述的“c6~c14的芳基”例如苯基、萘基、芴基或蒽基,又例如苯基)、或r1-3取代或未取代的c5~c10的环烷基(所述的r1-3的个数可为一个或多个[例如2个或3个],当存在多个r1-3时,任意两个r1-3相同或不同;所述的“c5~c10的环烷基”例如c5~c6的环烷基,又例如环戊基或环己基);
所述的r1-1、r1-2和r1-3各自独立地为:c1~c4烷基(例如甲基、乙基、丙基或丁基)或苯基;
m1和m2各自独立地为:ni或pd;
x1和x2各自独立地为:卤素(例如氟、氯、溴或碘,又例如氯或溴)、r2-1取代或未取代的c1~c10的烷基(所述的r2-1的个数可为一个或多个[例如2个或3个],当存在多个r2-1时,任意两个r2-1相同或不同;所述的“c1~c10的烷基”例如为c1~c6的烷基,又例如甲基、乙基、丙基、丁基或己基)、r2-2取代或未取代的c5~c10的环烷基(所述的r2-2的个数可为一个或多个[例如2个或3个],当存在多个r2-2时,任意两个r2-2相同或不同;所述的“c5~c10的环烷基”例如c5~c6的环烷基,又例如环戊基或环己基)、r2-3取代或未取代的苯基(所述的r2-3的个数可为一个或多个[例如2个或3个],当存在多个r2-3时,任意两个r2-3相同或不同;所有的r2-3可独立地位于“苯基与其他基团连接位点”的邻位、间位或对位,例如对位)、r2-4取代或未取代的苄基(所述的r2-4的个数可为一个或多个[例如2个或3个],当存在多个r2-4时,任意两个r2-4相同或不同;所有的r2-4可独立地位于“苄基中亚甲基”的邻位、间位或对位,例如对位)或2-位c1-c4烷基取代或者未取代的烯丙基(其中,所述的“c1-c4烷基”例如为甲基、乙基、丙基或丁基);
所述的r2-1、r2-2、r2-3和r2-4各自独立地为:c1~c4烷基(例如甲基、乙基、丙基或丁基)、苯基或-cf3;
y1、y2、y3和y4各自独立地为:r3-1取代或未取代的c1~c10的烷基(所述的r3-1的个数可为一个或多个[例如2个或3个],当存在多个r3-1时,任意两个r3-1相同或不同;所述的c1~c10的烷基例如c1~c6的烷基,又例如甲基、乙基、丙基、丁基或己基)、r3-2取代或未取代的c6~c14芳基(所述的r3-2的个数可为一个或多个[例如2个或3个],当存在多个r3-2时,任意两个r3-2相同或不同;所述的“c6~c14的芳基”例如苯基、萘基、芴基或蒽基,又例如苯基)、r3-3取代或未取代的“杂原子选自n、o和s中的一种或多种,杂原子数为1~3个的4~7元杂环烷基”(所述的r3-3的个数可为一个或多个[例如2个或3个];当存在多个r3-3时,任意两个r3-3相同或不同;所有的r3-3可独立地位于所述的“杂原子选自n、o和s中的一种或多种,杂原子数为1~3个的4~7元杂环烷基与其他基团连接位点”的邻位、间位或对位,例如邻位或对位;所述“杂原子选自n、o和s中的一种或多种,杂原子数为1~3个的4~7元杂环烷基”例如四氢呋喃基或四氢吡喃基)、r3-4取代或未取代的“杂原子选自n、o和s中的一种或多种,杂原子数为1~3个的4~6元杂芳基”(所述的r3-4的个数可为一个或多个[例如2个或3个],当存在多个r3-4时,任意两个r3-4相同或不同;所有的r3-4可独立地位于所述的“杂原子选自n、o和s中的一种或多种,杂原子数为1~3个的4~6元杂芳基”与其他基团连接位点的邻位、间位或对位,例如邻位或对位;所述的杂原子可为n和/或o;所述的杂原子数可为1个或2个;所述的杂芳基可为5元或6元;所述的“杂原子选自n、o和s中的一种或多种,杂原子数为1~3个的4~6元杂芳基”例如噻吩基、吡咯基或吡啶基);
所述的r3-1、r3-2、r3-3和r3-4各自独立地为:-or4、-sr5、-nr6r7、-pr8r9、-p(o)r10r11、-sir12、c1~c4烷基(例如甲基、乙基、丙基或丁基)、卤素(例如氟、氯、溴或碘,又例如氟或氯)、苯基或二苯基甲基(ph2ch-);
所述的r4、r5、r6、r7、r8、r9、r10、r11和r12各自独立地为:c1~c10的烷基(例如c1~c6的烷基,又例如甲基、乙基、丙基、丁基或己基)、c5~c10的环烷基-(ch2)m-(其中,m=0、1、2或3,例如0或1;所述的“c5~c10的环烷基”例如环戊基或环己基;所述的“c5~c10的环烷基-(ch2)m-”例如
所述的r4-1和r4-2各自独立地为:卤素(例如氟、氯、溴或碘,又例如氟或氯)、-cf3、c1~c4烷氧基(例如甲氧基、乙氧基、丙氧基或丁氧基)、或c1~c4烷基(例如甲基、乙基、丙基或丁基);
且所述的金属配合物i是电荷平衡的。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:所述的r3-1、r3-2、r3-3或r3-4为-or4时,所述的r4为c1~c10的烷基、或c5~c10的环烷基-(ch2)m-。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的r3-1、r3-2、r3-3或r3-4为-sr5时,所述的r5为c1~c10的烷基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的r3-1、r3-2、r3-3或r3-4为-nr6r7时,所述的r6或r7为c1~c10的烷基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的r4、r5、r6、r7、r8、r9、r10、r11或r12为c1~c10的烷基时,所述的c1~c10的烷基为甲基、乙基、丙基、丁基或己基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的r4、r5、r6、r7、r8、r9、r10、r11或r12为c5~c10的环烷基-(ch2)m-时,所述的c5~c10的环烷基-(ch2)m-为
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:所述的y1、y2、y3和y4各自独立地为
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:r1、r2、r4和r5各自独立地为:-cf3、r1-1取代或未取代的c1~c10的烷基、或r1-2取代或未取代的c6~c14芳基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:r3和r6各自独立地为氢、或r1-1取代或未取代的c1~c10的烷基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:x1和x2各自独立地为卤素、或r2-1取代或未取代的c1~c10的烷基或2-位甲基取代或者未取代的烯丙基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:所述的r4、r5、r6、r7、r8、r9、r10、r11和r12各自独立地为c1~c10的烷基、或c5~c10的环烷基-(ch2)m-。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:所述的r3-1、r3-2、r3-3和r3-4各自独立地为-nr6r7、-or4、-sr5、c1~c4烷基、卤素或苯基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:y1、y2、y3和y4各自独立地为r3-1取代或未取代的c1~c10的烷基、或r3-2取代或未取代的c6~c14芳基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的y1、y2、y3和y4各自独立地为r3-2取代或未取代的c6~c14芳基时,所述的r3-2为-or4、-sr5、c1-c4烷基、卤素或苯基。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的y1、y2、y3和y4各自独立地为r3-1取代或未取代的c1~c10的烷基时,所述的r3-1为-nr6r7。
在某一方案中,所述的金属配合物i的某些基团的定义如下,未定义的基团如前任一方案所述:当所述的y1、y2、y3或y4上含有杂原子时,所述的杂原子可任选地与m1或m2形成或不形成配位键。
在某一方案中,所述的金属配合物i可为如下任一化合物:
本发明提供了一种所述的金属配合物i的制备方法,其包括如下步骤:在有机溶剂中,在碱存在下,将化合物ii与金属试剂进行如下所示的络合反应,得到所述的金属配合物i,即可;所述的化合物ii为如式ii-a和ii-b所示化合物的混合物;所述的金属试剂为m1x1和/或m1x1·b,及m2x2和/或m2x2·b;其中b为配体或配位性溶剂;所述的化合物ii与所述金属试剂总量的摩尔比为1:0.8~1:1.2;
其中,所述的r1、r2、r3、r4、r5、r6、x1、x2、y1、y2、y3和y4均如上所述。
所述的化合物ii-a和化合物ii-b可相同或不同。
所述的m1x1·b和m2x2·b中,所述的配体或配位性溶剂b与m1或m2形成配位键。所述的配体或配位性溶剂b可为本领域中常规的,例如“含杂原子n、o、p和s中的一种或多种”的、且与第八族金属m可以形成配位键的配体或溶剂;本发明中所述的配位性溶剂b较佳地为乙醇、甲醇、乙二醇、乙腈、乙二醇二甲醚(dme)、四氢呋喃、乙酰丙酮(acac)、吡啶、n,n-二甲基甲酰胺(dmf)和水中的一种或多种;本发明中所述的配体b较佳地为三苯基膦。
较佳地,本发明中所述的m1x1·b和m2x2·b各自独立地为nibr2(dme),nime2py4,niphclpph3,nimeclpph3,nicl2·2h2o或nicl2·4ch3ch2oh(又例如nibr2(dme)或nime2py4)。
本发明中所述的m1x1或m2x2较佳地为pdcl2。
所述的有机溶剂可为本领域中常规的有机溶剂,以用了不影响反应的进行即可,例如弱极性有机溶剂;本发明中较佳地为醚类溶剂(例如四氢呋喃和/或乙醚)、芳香烃类溶剂(例如甲苯)、烷基类溶剂(例如己烷)、乙腈和卤代烃类溶剂(例如二氯甲烷)中的一种或多种。
本发明中,化合物ii与金属试剂的比例对金属配合物i的制备至关重要,化合物ii与金属试剂总量的比例控制在0.8-1.2为宜。在惰性溶剂中,用式ii化合物分步与0.4-0.6当量的m1x12(或m1x12·b)以及0.4-0.6当量的m2x22(或m2x22·b)反应,得到式i配合物;或用式ii化合物直接与0.8-1.2当量的m1x12(或m1x12·b)或0.8-1.2当量的m2x22(或m2x22·b)反应,得到式i配合物。
所述的化合物ii与所述的金属试剂总量的摩尔比较佳地为1:1。
所述的碱可为本领域该类反应中常规的碱,例如,kh、nah、buli、et3n和吡啶中的一种或多种,较佳地为kh和/或nah。
所述碱的用量可为本领域该类反应中常规的,本发明中,所述的碱与所述的化合物ii的摩尔比可为1:1~5:1;较佳地为1.2:1~4:1(例如2:1)。
所述络合反应的温度可为本领域该类反应中常规的,例如-50℃~100℃(-30℃~30℃)。
所述的络合反应的进程可采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行监测,一般以化合物ii不再反应时为反应终点,反应时间可为2h~48h(例如16~24h)。
所述的金属配合物i的制备方法,其还可包括如下步骤:在有机溶剂中,将化合物iii-a与含[r3]-基团的亲核试剂、或化合物iii-b与含[r6]-基团的亲核试剂进行如下所示的反应,分别得到所述的化合物ii-a或化合物ii-b即可;当[r3]-或[r6]-基团为h时,为还原反应;当[r3]-或[r6]-基团不为h时,为羰基加成反应;
其中,r1、r2、r3、r4、r5、r6、y1、y2、y3和y4均如上所述。
所述的还原反应或羰基加成反应的反应条件可为本领域该类反应中常规的反应条件。
所述的亲核试剂可为本领域中该类反应中常规的,例如当[r3]-或[r6]-基团为h时,所述的亲核试剂为金属氢化物(例如氢化钠和/或氢化钾);当[r3]-或[r6]-基团不为h时,所述的亲核试剂为丁基锂(buli)、烷基铝(alet3、alme3、et2alcl、etalcl2、me2alcl或al(i-bu)3)和格氏试剂(例如etmgbr、memgbr或bumgbr)中的一种或多种。
所述的有机溶剂可为本领域中常规的有机溶剂,以用了不影响反应的进行即可,例如弱极性有机溶剂;本发明中较佳地为醚类溶剂(例如四氢呋喃、乙醚)、芳香烃类溶剂(例如甲苯)、烷基类溶剂(例如己烷)、乙腈和卤代烃类溶剂(例如二氯甲烷)中的一种或多种。
所述的化合物ii-a或化合物iii-b与所述的亲核试剂的摩尔比可为本领域该类反应中常规的,例如1:1~1:2.0。
所述反应的温度可为本领域该类反应中常规的,例如-50℃~100℃(-30℃~30℃)。
所述的化合物iii(iii-a和iii-b)可参照专利cn201410412200.8的制备方法制备得到。
本发明提供了一种如上所述的金属配合物i在聚合反应中的应用。
本发明提供了一种乙烯聚合物的制备方法,在如上所述的金属配合物i单独或所述的金属配合物i与助催化剂w存在下,将乙烯进行均聚合反应,得到所述的乙烯聚合物,即可。
其中,所述的均聚合反应的反应条件可为本领域该类反应中常规的反应条件;例如为如下反应条件:
所述的助催化剂w可为本领域该类反应中常规的助催化剂,例如烷基铝化合物(例如alet3、alet2cl、aletcl2、alme2cl、alme3和al(i-bu)3中的一种或多种)、烷基铝氧烷[例如甲基铝氧烷(例如购自于akzonobel的产品mao,1.5m(mol/l)的甲苯溶液)、改性的/修饰的甲基铝氧烷(例如购自于akzonobel的产品mmao,2.0m(mol/l)的正庚烷溶液)、乙基铝氧烷和丁基铝氧烷中的一种或多种]、和含弱配位阴离子的金属盐(例如na[b(3,5-(cf3)2c6h3)4]和/或agoso2cf3)中的一种或多种。
所述的金属配合物i与所述的助催化剂w的摩尔比可为本领域中常规的,例如1:1~1:10000(又例如1:100、1:1500、1:1000、1:2000、1:2500、1:3000、1:4000或1:5000)。
本发明中较佳地等当量或略多些的烷基铝预先与极性单体接触,从而形成对催化剂的保护;在聚合过程中再加入大量的烷基铝作为助催化剂。
所述的均聚合反应没有特别限制,可以采用任何本领域的常规技术,如淤浆聚合、环管聚合、气相聚合,或其他形式的常规聚合反应工艺。
所述的均聚合反应一般在惰性溶剂中进行,例如烃类、环烃类或芳烃类。为有利于反应器操作与聚合产物,惰性溶剂的种类没有特别限制,可使用小于12个碳的烃类,例如丙烷、异丁烷、正戊烷、2-甲基丁烷、己烷、甲苯和氯苯中的一种或多种。
所述均聚合反应的温度没有特别限制,根据聚合反应的种类、设备、目标产物而选择。较佳地,所述均聚合反应的温度可维持在20至150℃,为达到好的催化活性与生产能力,可维持在20至120℃(例如30℃)。
所述均聚合反应的压力也没有特别限制,可根据均聚合反应的设备、目标产物而在0.1至10mpa内变化,较佳地在0.1~5mpa内变化。在本发明的一个优选例中,在0.1至3mpa内操作可获得较好的反应器操作参数与聚合物。
本发明提供了一种乙烯聚合物,通过如上所述的制备方法制得。
本发明提供了一种乙烯聚合物,其具有选自下组的一个或多个特征:
a)数均分子量为600,000至2000,000g/mol,分子量分布pdi=1.5~3.0;
b)均聚物支链化程度为1至20个支链/1000碳,所述的烷烃支链包含甲基支链、乙基支链、丙基支链以及大于c4的支链。所述的烷烃支链中甲基支链比例0.75-0.95,乙基支链比例0.01-0.05,丙基支链比例0.01-0.05,大于c4的支链比例0.01-0.10。
c)共聚物熔融温度tm=70~140℃。
在另一优选例中,所述的乙烯均聚物具有下组一个或多个特征:
a)数均分子量为1000,000至1400,000g/mol,pdi=1.5~2.5;
b)均聚物支链化程度为1至20个支链/1000碳,所述的烷烃支链包含甲基支链、乙基支链、丙基支链以及大于c4的支链。所述的烷烃支链中甲基支链比例0.85-0.95,乙基支链比例0.01-0.03,丙基支链比例0.01-0.03,大于c4的支链比例0.01-0.05。
c)共聚物熔融温度tm=100~140℃。
在另一优选例中,所述的乙烯均聚物具有下组一个或多个特征:
a)数均分子量为1200,000至200,000g/mol,pdi=1.5~2.5;
b)均聚物支链化程度为1至10个支链/1000碳,所述的烷烃支链包含甲基支链、乙基支链、丙基支链以及大于c4的支链。所述的烷烃支链中甲基支链比例0.85-0.95,乙基支链比例0.01-0.02,丙基支链比例0.01-0.02,大于c4的支链比例0.01-0.03。
c)共聚物熔融温度tm=130~140℃。
术语
如本文所用,“助催化剂w”指能够与本发明的催化剂一同用于催化聚合反应,且能够改善反应的物质。在本发明中,优选的助催化剂w可以是中性的路易斯酸(lewisacid);可从金属m拔去x-形成(wx)-;当(wx)-是弱配位的阴离子时,w可将烷基或氢转移到金属m上,如烷基铝化合物尤其是甲基铝氧烷(实施例中简写为mao)或改性的甲基铝氧烷(实施例中简写为mmao);或者也可组合使用两种化合物,其中一种可将烷基或氢转移到金属m上,如烷基铝化合物(尤其是alet3、alme3或al(i-bu)3);另一种可从金属m拔去x-形成弱配位阴离子,如钠盐或银盐(例如na[b(3,5-(cf3)2c6h3)4]或agoso2cf3)。
术语“弱配位的阴离子”是指相对不配位的阴离子,其配位状况可参见文献(w.beck.,etal.,chem.rev.,vol.88,p1405-1421(1988),和s.h.stares,chem.rev.,vol.93,p927-942(1993))及其参考文献,例如(r14)3alx-、(r14)2alx2-、(r14)alx3-、sbf6-、pf6-、bf4-、(c6f5)4b-、(rfso2)2,n-、cf3so3-、或((3,5-(cf3)2)c6h3)4b-。
在各个化合物结构式中,“→”表示配位键。
本发明的积极进步效果在于:(1)本发明提供的化合物可作为一类新的烯烃聚合催化剂,能够被用于制备高分子量的烯烃聚合物。
(2)本发明提供的化合物可单独或在助催化剂的作用下,催化乙烯均聚合,催化活性高(可达>106g/(molmetal)·h),所制备聚乙烯具有高分子量高(可达mn=105~106g/mol)、分子量分布较窄(pdi=1.0~2.5)的特点;此外,制备的聚乙烯含有烷烃支链(1~20支链/1000碳),所述的烷烃支链包含甲基支链、乙基支链、丙基支链以及大于c4的支链。所述的烷烃支链中甲基支链比例为0.75-0.95,乙基支链含量0.01-0.05,丙基支链含量0.01-0.05,大于c4的支链含量0.01-0.10;聚合物熔融温度tm=70~140℃。
附图说明
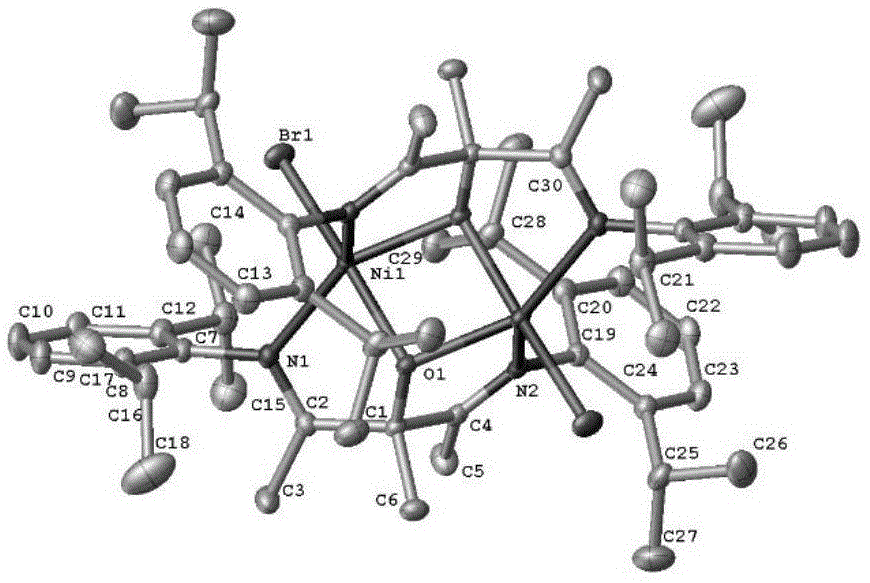
图1为金属配合物i-1的单晶结构;
图2为金属配合物i-6的单晶结构;
图3为金属配合物i-7的单晶结构;
图4为实施例6中金属配合物i-6催化乙烯聚合的聚合物gpc图;
图5为实施例6中金属配合物i-7催化乙烯聚合的聚合物gpc图;
图6为实施例7中金属配合物i-6催化乙烯聚合的聚合物gpc图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
下述实施例中,助催化剂用量单位为mmol;极性单体用量单位为mmol;极性单体插入率单位为mol%;聚合温度单位为℃;tm单位为℃;branch:支化度,单位为支链/1000c;聚合活性单位为105mol·h·atm。支链比例为所述支链在总烷基支链中的质量百分比。mn:数均分子量;mw:重均分子量;mw/mn:分子量分布。
甲基铝氧烷(mao)购自于akzonobel的产品,1.5m(mol/l)的甲苯溶液)。
改性的/修饰的甲基铝氧烷(mmodifiedmao,mmao)购自于akzonobel的产品,2.0m(mol/l)的正庚烷溶液。
除特别说明外,所有反应均在氮气氛围中进行。所有原料及溶剂除了注明外,均为商品化试剂。其中氢化钾经正己烷洗净抽干后保存在手套箱中备用。甲苯、thf、乙腈、二氯甲烷以及正己烷用mbraunsps-800除水除氧装置处理后使用。
有机反应均使用tlc跟踪或核磁跟踪,使用烟台化工研究所生产的高效薄层层析板。快速柱层析使用烟台化工研究所生产的h型硅胶。1hnmr、13cnmr、19fnmr和31pnmr在varian400-mr(400mhz)、agilent400mrdd2(400mhz)以及agilent600mrdd2(600mhz)型核磁共振仪上测定。x-射线晶体衍射在brukersmartccd或rigakuafc7r或brukerd8venture上测定,具体方法是:在氮气保护下将单晶封在毛细管中,在石墨单色mo-ka或ga-ka射线照射下,用brukersmartccd或rigakuafc7r或brukerd8venture衍射仪收集晶体的衍射强度数据。
聚合物分子量mw、mn以及分子量分布(mw/mn)利用watersalliancegpc2000在1,2,4-三氯苯(流速1.0ml/min)中135℃下,以聚苯乙烯为标样测定。
聚合物熔点测定在perkin-elmerpyris1型差热扫描量热分析仪(dsc)上进行,升温速率为5℃/min,温度范围为20~200℃,为了消除热历史,同样的扫描参数进行两次扫描,实验结果取第二次扫描的结果。研究不同聚合物的熔点。
聚合物1h-nmr谱在varianxl-400mhz核磁共振仪上以d4-o-二氯苯为溶剂,在110℃下测定。共聚单体插入率由1h-nmr谱提供的信息计算得到。
式iii所示的化合物参照文献步骤合成(yokota,s.;tachi,y.;itoh,s.inorg.chem.2002,41,1342)。
聚合物支化度通过高温定量氢谱确定:以氘代四氯乙烷为氘代试剂在110℃下溶解聚合物,该聚合物样品通过核磁在110℃时测定定量氢谱。0.93ppm处单峰或多峰为支链末端甲基(-ch3)的信号峰,设此峰面积为3,1.35ppm处的单峰为聚合物共聚单元中亚甲基(-ch2-)的信号峰,峰面积积分为n,则支链化程度(x支链/1000c)通过以下公式计算:
x=2000÷(n+2)(公式1-5)
甲基支链、乙基支链、丙基支链、仲丁基支链以及大于c4的支链比例通过高温氢谱结合高温碳谱确定:以氘代四氯乙烷为氘代试剂在110℃下溶解聚合物,该聚合物样品通过核磁在110℃时测定定量碳谱。其中,20.26ppm处的信号峰为甲基的信号峰,该峰积分面积定为a;11.45ppm处的信号峰为乙基的信号峰,该峰积分面积定为b;14.90ppm处的信号峰为丙基的信号峰,该峰积分面积定为c;11.69,19.74ppm处两组信号峰为仲丁基基的信号峰,
则:甲基支链比例为a÷(a+b+c+d+e);
乙基支链比例为b÷(a+b+c+d+e);
丙基支链比例为c÷(a+b+c+d+e);
仲丁基基支链比例为d÷(a+b+c+d+e);
大于c4的支链比例为e÷(a+b+c+d+e)。
实施例1式ii化合物的合成
化合物ii-1的制备方法如下所示:
12.5g(125mmol)乙酰丙酮与44.3g(250mmol)2,6-二异丙基苯胺在10.6ml(125mmol)浓盐酸作用下,即可顺利以81%的产率得到44.2g化合物vi-1,该化合物在1当量醋酸铜作用下通氧气氧化得到化合物iii-1,随后通过alme3加成得到产物ii-1,三步总产率65%,共36g。
化合物ii-11的制备方法如下所示:
将19.4g(193.8mmol)乙酰丙酮和33.8g(190.87mmol)2,6-二异丙基苯胺溶于100ml甲苯中,并加入约1.0g(2%)的一水合对甲苯磺酸,升温回流分水至无水分出。然后减压下除去溶剂,残留物减压蒸馏即可以83%的产率得到黄色油状物v-1141.3g。
将3.4g(38.7mmol)n,n-二甲基乙二胺与8g(30.7mmol)v-11在0.15g一水合对甲苯磺酸下作用下即可顺利以81%产率得到8.2g化合物iv-11,该化合物在1当量醋酸铜作用下通氧气氧化得到化合物iii-11,随后通过alme3加成反应得到产物ii-11,三步总产率72%,共7.2g。
化合物ii-19的制备方法如下所示:
将19.4g(193.8mmol)乙酰丙酮和17.8g(190.87mmol)苯胺溶于100ml甲苯中,并加入约1.0g(2%)的一水合对甲苯磺酸,升温回流分水至无水分出。然后减压下除去溶剂,残留物减压蒸馏即可以78%的产率得到黄色油状物v-1926.5g。
将5.0g(38.7mmol)2,6-二氟苯胺与5.4g(30.7mmol)v-19在0.15g一水合对甲苯磺酸下作用下即可顺利以78%产率得到6.8g化合物vi-19,该化合物在1当量醋酸铜作用下通氧气氧化得到化合物iii-19,随后通过lialh4还原反应得到产物ii-19,三步总产率56%,共4.1g。
化合物ii-2~ii-10、ii-12~ii-22的合成
按照以上制备ii-1的实验方法,使用相应的原料,制备得到其它化合物:ii-2(产率64%);ii-3(产率75%);ii-4(产率51%);ii-5(产率61%);ii-13(产率87%);ii-15(产率82%);ii-20(产率67%);ii-22(产率68%);
按照以上制备ii-11的实验方法,使用相应的原料,得到其它化合物:ii-6(产率41%);ii-7(产率45%);ii-8(产率56%);ii-9(产率61%);ii-10(产率51%);ii-12(产率75%);ii-14(产率52%);ii-16(产率43%);ii-17(产率63%);ii-18(产率55%);
按照以上制备ii-19的实验方法,使用相应的原料,得到化合物ii-21(产率57%)。
部分化合物的核磁数据(1hnmr)如下:
ii-1
1hnmr(400mhz,cdcl3):δ=7.18-7.09(m,6h,ar-h),6.29(brs,1h,oh),2.79-2.65(m,4h,ch(ch3)2),1.86(s,6h,ch3),1.83(s,3h,ch3),1.19-1.16(m,24h,ch3).
ii-3
1hnmr(400mhz,cdcl3):δ=7.05-7.03(s,4h,b,hm),6.94-6.91(2h,b,hp),6.16(brs,1h,oh),2.02(s,6h,o-ch3),1.99(s,6h,o-ch3),1.83(s,6h,ch3),1.81(s,3h,ch3).
ii-4
1hnmr(400mhz,cdcl3):δ=7.10-7.03(m,4h,ar-h),6.22(brs,1h,oh),2.43-2.34(m,8h,ch2ch3),1.84(s,6h,ch3),1.82(s,3h,ch3),1.15(t,24h,ch2ch3).
ii-6
1hnmr(400mhz,cdcl3):δ=6.71(s,4h,ar-h),6.35(brs,1h,oh),3.83(s,6h,och3),2.74-2.64(m,4h,ch(ch3)2),1.83(s,6h,ch3),1.80(s,3h,ch3),1.14(m,24h,ch(ch3)2).
ii-7
1hnmr(400mhz,cdcl3):δ=7.09(s,4h,ar-h),6.10(brs,1h,oh),2.74-2.64(m,4h,ch(ch3)2),1.81(s,6h,ch3),1.77(s,3h,ch3),1.12(t,24h,ch(ch3)2).
实施例2金属化合物i-1的合成
将化合物ii-1(0.67g,1.5mmol)的四氢呋喃溶液于室温下缓慢滴加至kh(0.12g,3.0mmol)的四氢呋喃中,滴加过程中有大量气泡产生。十分钟内滴加完毕,反应12小时后,透过硅藻土过滤。室温下将滤液缓慢滴加至nibr2(dme)(0.46g,1.5mmol)的四氢呋喃溶液中,反应过夜。减压除去溶剂,加入甲苯,过滤除去kbr,滤液浓缩并加入少量己烷重结晶,得到红色固体i-10.46g,产率52%。
元素分析:实测(计算值):c:60.98(61.46);h:7.59(7.39);n:4.59(4.78)。
实施例3金属化合物i-2~i-22的合成
用与实施例2相同的实验方法得到其它化合物i-2(产率65%);i-3(产率71%);i-4(产率59%);i-5(产率63%);i-6(产率74%);i-7(产率76%);i-8(产率64%);i-9(产率67%);i-10(产率55%);i-11(产率55%);i-12(产率75%);i-13(产率72%);i-14(产率58%);i-15(产率66%);i-16(产率61%);i-17(产率72%);i-18(产率54%);i-19(产率67%);i-20(产率77%);i-21(产率68%);i-22(产率71%)。
各原料和产物的对应关系见下表。
金属化合物i-2~i-22的结构如下:
各化合物的元素分析数据如下:
i-2元素分析:实测(计算值):c:38.84(38.90);h:2.85(2.72);n:5.15(5.04)。
i-3元素分析:实测(计算值):c:56.01(55.74);h:5.79(5.74);n:6.20(5.91)。
i-4元素分析:实测(计算值):c:58.63(58.90);h:6.55(6.65);n:5.39(5.28)。
i-5元素分析:实测(计算值):c:52.30(51.90);h:5.38(5.37);n:4.36(4.04)。
i-6元素分析:实测(计算值):c:59.00(59.47);h:7.25(7.33);n:4.01(4.33)。
i-7元素分析:实测(计算值):c:55.12(55.00);h:6.25(6.31);n:5.01(4.28)。
i-8元素分析:实测(计算值):c:57.00(57.41);h:6.42(6.22);n:5.61(5.58)。
i-9元素分析:实测(计算值):c:56.72(56.43);h:6.32(6.25);n:5.30(5.26)。
i-10元素分析:实测(计算值):c:60.94(60.61);h:7.12(7.06);n:4.68(4.56)。
i-11元素分析:实测(计算值):c:53.00(53.15);h:7.41(7.30);n:8.62(8.45)。
i-12元素分析:实测(计算值):c:55.12(54.89);h:7.94(7.68);n:8.22(8.00)。
i-13元素分析:实测(计算值):c:46.27(46.58);h:8.28(8.04);n:12.12(12.07)。
i-14元素分析:实测(计算值):c:60.81(60.76);h:6.43(6.34);n:4.29(4.29)。
i-15元素分析:实测(计算值):c:56.58(56.60);h:5.76(5.99);n:5.53(5.74)。
i-16元素分析:实测(计算值):c:44.55(44.41);h:3.62(3.52);n:5.99(5.75)。
i-17元素分析:实测(计算值):c:60.56(60.25);h:7.13(6.76);n:4.96(5.21)。
i-18元素分析:实测(计算值):c:54.72(54.83);h:5.22(5.48);n:6.55(6.09)。
i-19元素分析:实测(计算值):c:46.31(46.42);h:3.29(3.44);n:6.45(6.37)。
i-20元素分析:实测(计算值):c:44.52(44.41);h:3.62(3.37);n:5.01(4.93)。
i-21元素分析:实测(计算值):c:62.70(62.63);h:4.78(4.53);n:5.30(5.04)。
i-22元素分析:实测(计算值):c:53.01(53.50);h:4.71(4.96);n:4.76(5.12)。
实施例4金属化合物i-23的合成
在100ml的反应瓶中,加入4.49g(10.0mmol)化合物前体,用20ml四氢呋喃溶解,-30℃滴加至0.29g(12.0mmol)氢化钠的四氢呋喃(10ml)中,室温反应24h后,将钠盐滴加至pdcl2(10.0mmol)的四氢呋喃(10ml)中,室温反应24h,停止反应,减压除去溶剂,加入20ml甲苯,加热溶解,过滤,母液处于-30℃冷冻,即得到产物,干燥后得到墨绿色晶体i-23,4.2g(产率75%)。
元素分析:实测(计算值):c:61.36(61.12);h:7.30(7.35);n:5.02(4.75)。
实施例5金属配合物i-24的合成
在100ml的反应瓶中,加入4.49g(10.0mmol)化合物前体,用20ml四氢呋喃溶解,-30℃滴加至0.29g(12.0mmol)氢化钠的四氢呋喃(10ml)中,室温反应24h后,将钠盐滴加至nime2py4(10mmol)的四氢呋喃(10ml)中,室温反应24h,停止反应,减压除去溶剂,加入20ml甲苯,加热溶解,过滤,母液处于-30℃冷冻,即得到产物,干燥后得到绿色晶体i-24,4.1g(产率80%)。
元素分析:实测(计算值):c:71.63(71.41);h:8.93(8.89);n:5.43(5.37)。
实施例6催化剂i-1~i-10催化乙烯聚合实验
以如式a所示的催化剂作对比进行如下实验;催化剂a参照文献:azoulay,j.d.;rojas,r.s.;serrano,a.v.;ohtaki,h.;galland,g.b.;wu,g.;bazan,g.c.angew.chem.int.ed.2009,48,1089.制备得到。
在0.1mpa的乙烯气氛下,依次将甲苯100ml、etalcl2(etalcl2与催化剂的摩尔比为500)加入经干燥的250ml的聚合瓶中,搅拌,然后置于30℃油浴中,恒温一定时间,加入催化剂i-1~i-10及a(5μmol)的甲苯溶液,反应10分钟后,用含5%盐酸的乙醇终止反应。聚合物经沉淀、过滤、洗涤后,于50℃真空干燥至恒重得聚乙烯。乙烯均聚结果如下表。
催化剂i-1~i-10催化乙烯聚合结果
注:cat:催化剂;w:聚乙烯重量;activity:活性;mw:重均分子量;mw/mn:分子量分布;branch:支化度(即聚乙烯链中每1000个碳含有支链的数目)。
结果显示,该类催化剂体系在常压下即可催化乙烯聚合,活性可达106g/mol·h,数均分子量为105g/mol,支化度为21-26.2/1000c。
实施例7部分催化剂高压下催化乙烯聚合实验
在1.0mpa的乙烯气氛下,依次将甲苯100ml、etalcl2(etalcl2与催化剂的摩尔比为1500)加入经干燥的350ml的高压釜中,剧烈搅拌,然后置于30℃油浴中,恒温一定时间,加入催化剂i-1、i-2、i-3、i-4、i-5的甲苯溶液(各5μmol),反应10分钟后,用含5%盐酸的乙醇终止反应。聚合物经沉淀、过滤、洗涤后,于50℃真空干燥至恒重得乙烯聚合物。乙烯聚合结果如下表。
部分催化剂高压下催化乙烯聚合的结果
注:cat:催化剂;w:聚乙烯重量;activity:活性;mw:重均分子量;mw/mn:分子量分布;branch:支化度(即聚乙烯链中每1000个碳含有支链的数目)。
结果显示,相对于常压聚合,得到的聚合物数均分子量可达106g/mol,支化度降低。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!