邻位二苯甲基取代的亚胺膦镍、钯配合物、制备方法与应用与流程
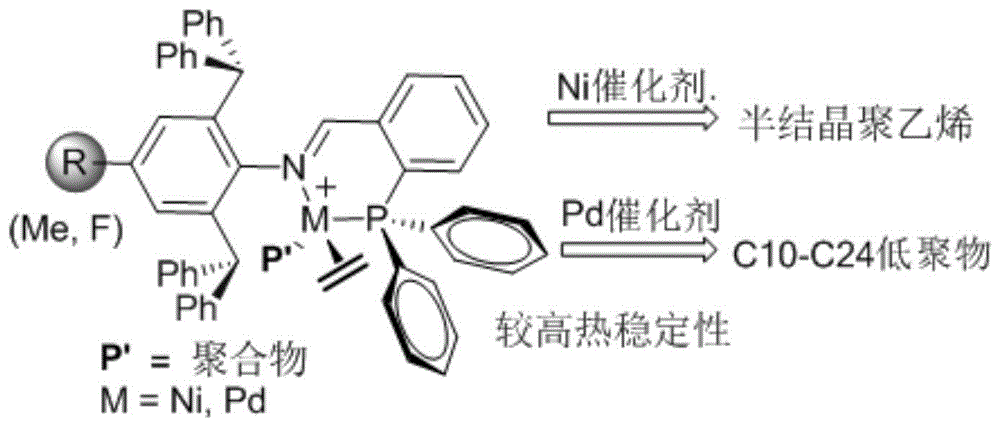
本发明涉及烯烃聚合技术领域,具体涉及一种邻位二苯甲基取代的亚胺膦镍、钯配合物、制备方法与应用。
背景技术:
聚烯烃因其原料丰富、价格低廉,成为了目前世界上产量最大、用途最为广泛、最为重要的高分子材料。种类繁多的聚烯烃产品作为材料有着不可多得的优点,例如材料力学性能和热性能调变范围大,优良的加工性能,稳定可靠,可循环再利用等优点,广泛应用于农业,军事,科研,日常生活等许多领域,与国民经济和人民生活密切相关。聚烯烃的蓬勃发展使得新型烯烃聚合催化剂的开发和研究成为人们关注的热点。
后过渡金属催化剂是对烯烃聚合具有高活性的新一代金属有机催化剂。近年来,镍钯催化剂催化乙烯和α-烯烃的链行走聚合被广泛研究。特别是大量修饰brookhart二亚胺催化剂(f.z.wang,c.l.chen,acontinuinglegend:thebrookhart-typeα-diiminenickelandpalladiumcatalysts.polym.chem.2019,10,2354–2369.),通过配体骨架,位阻和电子效应调控来精确的控制高分子拓扑结构。很多研究证明,大体积的二苯甲基基团引入配体中n-芳环的邻位,可以有效地提高镍和钯配合物的催化性能。而对位含芳基取代的α-二亚胺镍配合物催化乙烯聚合得到较高支化的无定形聚乙烯,可塑性较差。近几年,在乙烯与极性单体共聚中,一系列基于二苯基膦的钯镍催化剂与α-亚胺相比具有相似甚至更好的性能。早期,邻位异丙基取代的亚胺膦镍催化剂催化乙烯聚合(g.l.crossetti,m.l.dias,b.t.queiroz,l.p.silva,c.m.ziglio,j.a.s.bomfim,c.a.l.filgueiras,ethylenepolymerizationwithimineandphosphinenickelcomplexescontainingisothiocyanate.appl.organometal.chem.2004,18,331–336.),表现出很低的催化活性,并没有对其产品的结构进行分析。
技术实现要素:
本发明所要解决的技术问题之一在于提供一种较高催化活性的邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物。
本发明通过以下技术手段实现解决上述技术问题的:
邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物,其结构式如下所示:
有益效果:邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物在亚胺氮原子芳环的邻位引入大体积基团二苯甲基,加速了乙烯插入的进程,提高了催化剂的活性和热稳定性,减少了活性链向单体的转移速率,得到高分子量聚合物。
优选的,所述邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物的制备方法包括以下步骤:
(1)制备邻位二苯甲基取代的亚胺膦配体
以乙醇为溶剂,以甲酸为催化剂,将邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛混合,于45~55℃下反应后,除去溶剂,制得邻位二苯甲基取代的亚胺膦配体;
(2)制备邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物
在惰性气体保护下,以二氯甲烷为溶剂,将步骤(1)中制得的邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的溴化镍混合,在室温下搅拌反应后,过滤悬浮液,母液在真空条件下除去溶剂后,制得邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物。
优选的,所述步骤(1)中邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛的摩尔比为1:1。
优选的,所述步骤(1)中反应的时间为12~24h。
优选的,所述步骤(1)中的反应时间为18h。
优选的,所述步骤(1)中邻位二苯甲基取代的苯胺为4-甲基-2,6-二苯甲基苯胺或4-氟-2,6-二苯甲基苯胺。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的溴化镍的摩尔比为1:1~1:1.2。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的溴化镍的摩尔比为1:1.1。
优选的,所述步骤(2)中被乙二醇二甲醚活化的溴化镍为nibr2(dme)。
优选的,所述步骤(2)中在室温下搅拌反应12~24h。
优选的,所述步骤(2)中在室温下搅拌反应18h。
本发明所要解决的技术问题之二在于提供一种具有较高催化活性的邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物的制备方法。
本发明通过以下技术手段实现解决上述技术问题的:
邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物的制备方法,包括以下步骤:
(1)制备邻位二苯甲基取代的亚胺膦配体
以乙醇为溶剂,以甲酸为催化剂,将邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛混合,于45~55℃下反应后,除去溶剂,制得粗产品,纯化后制得邻位二苯甲基取代的亚胺膦配体;
(2)制备邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物
在氮气保护下,以二氯甲烷为溶剂,将步骤(1)中制得的邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的溴化镍[nibr2(dme)]混合,在室温下搅拌反应后,过滤悬浮液,母液在真空条件下除去溶剂后,制得粗产品,纯化后制得邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物。
有益效果:本发明制得的邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物在亚胺氮原子芳环的邻位引入大体积基团二苯甲基,加速了乙烯插入的进程,提高了催化剂的活性和热稳定性,减少了活性链向单体的转移速率,得到高分子量聚合物。
优选的,所述步骤(1)中邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛的摩尔比为1:1。
优选的,所述步骤(1)中反应的时间为12~24h。
优选的,所述步骤(1)中反应的时间为18h。
优选的,所述步骤(1)中邻位二苯甲基取代的亚胺膦配体纯化方法包括以下步骤:将制得的粗产品用c2h5oh/ch2cl2混合溶剂重结晶析出固体沉淀,过滤干燥,制得邻位二苯甲基取代的亚胺膦配体。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的溴化镍的摩尔比为1:1~1.2。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的溴化镍的摩尔比为1:1.1。
优选的,所述步骤(2)中被乙二醇二甲醚活化的溴化镍为nibr2(dme)。
优选的,所述步骤(2)中在室温下搅拌反应12~24h。
优选的,所述步骤(2)中在室温下搅拌反应18h。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物的纯化方法包括以下步骤:将制得的粗产品用乙醚洗涤,真空干燥,制得邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物。
本发明所要解决的技术问题之三在于提供邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物在乙烯聚合中的应用。
本发明通过以下技术手段实现解决上述技术问题的:邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物在乙烯聚合中的应用。
有益效果:在助催化剂氯化二乙基铝的活化下催化乙烯,催化剂催化活性达到1.90×105gofpe(molni)-1h-1,表现出显著的热稳定性,并且提高了聚合活性和分子量,得到低度支化(28个支链/1000c)的半结晶聚乙烯,制得的聚合物具有较好的可塑性,可作为弹性体。
优选的,所述邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物用于乙烯聚合的方法包括以下步骤:将邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物与助催化剂以1:100~1000的摩尔比组成复合催化体系,催化乙烯聚合,得到乙烯聚合物。
优选的,所述催化体系中反应温度为20~80℃,反应时间为10~120min,邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物用量为2~10μmol/l。
本发明所要解决的技术问题之四在于提供一种具有较高催化活性的邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物。
本发明通过以下技术手段实现解决上述技术问题的:
邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物,其结构式如下所示:
有益效果:邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物在亚胺氮原子芳环的邻位引入大体积基团二苯甲基,加速了乙烯插入的进程,提高了催化剂的活性和热稳定性,减少了活性链向单体的转移速率,得到高分子量聚合物,可有效催化乙烯齐聚反应,得到高产率的油状低聚物,其结构主要以c10–c24的成分。
优选的,所述邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物的制备方法包括以下步骤:
(1)制备邻位二苯甲基取代的亚胺膦配体
以乙醇为溶剂,以甲酸为催化剂,将邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛混合,于45~55℃下反应后,除去溶剂,制得粗产品,纯化后制得邻位二苯甲基取代的亚胺膦配体;
(2)制备邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物
在惰性气体保护下,以二氯甲烷为溶剂,将含有邻位二苯甲基取代的亚胺膦配体与被乙腈活化的氯化钯混合,在室温下搅拌反应后,过滤悬浮液,母液在真空条件下除去溶剂后,制得粗产品,纯化后制得邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物。
优选的,所述步骤(1)中邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛的摩尔比为1:1。
优选的,所述步骤(1)中反应的时间为12~24h。
优选的,所述步骤(1)中反应的时间为18h。
优选的,所述步骤(1)中邻位二苯甲基取代的亚胺膦配体纯化方法包括以下步骤:将制得的粗产品用c2h5oh/ch2cl2混合溶剂重结晶析出固体沉淀,过滤干燥,制得邻位二苯甲基取代的亚胺膦配体。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的氯化钯的摩尔比为1:1~1.2。
优选的,所述步骤(2)中被乙腈活化的氯化钯为(ch3cn)2pdcl2。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物的纯化方法包括以下步骤:将制得的粗产品用乙醚洗涤,真空干燥,制得邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物。
本发明所要解决的技术问题之五在于提供一种具有较高催化活性的邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物的制备方法。
本发明通过以下技术手段实现解决上述技术问题的:
邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物的制备方法,包括以下步骤:
(1)制备邻位二苯甲基取代的亚胺膦配体
以乙醇为溶剂,以甲酸为催化剂,将邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛混合,于45~55℃下反应后,除去溶剂,制得粗产品,纯化后制得邻位二苯甲基取代的亚胺膦配体;
(2)制备邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物
在惰性气体保护下,以二氯甲烷为溶剂,将含有邻位二苯甲基取代的亚胺膦配体与被乙腈活化的氯化钯混合,在室温下搅拌反应后,过滤悬浮液,母液在真空条件下除去溶剂后,制得粗产品,纯化后制得邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物。
有益效果:本发明制得的邻位二苯甲基取代的亚胺膦配合物在亚胺氮原子芳环的邻位引入大体积基团二苯甲基,加速了乙烯插入的进程,提高了催化剂的活性和热稳定性,减少了活性链向单体的转移速率,得到高分子量聚合物,可有效催化乙烯齐聚反应,得到高产率的油状低聚物,其结构主要以c10–c24的成分。
优选的,所述步骤(1)中邻位二苯甲基取代的苯胺与邻二苯基膦苯甲醛的摩尔比为1:1。
优选的,所述步骤(1)中反应的时间为12~24h。
优选的,所述步骤(1)中反应的时间为18h。
优选的,所述步骤(1)中邻位二苯甲基取代的亚胺膦配体纯化方法包括以下步骤:将制得的粗产品用c2h5oh/ch2cl2混合溶剂重结晶析出固体沉淀,过滤干燥,制得邻位二苯甲基取代的亚胺膦配体。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦配体与被乙二醇二甲醚活化的氯化钯的摩尔比为1:1~1.2。
优选的,所述步骤(2)中被乙腈活化的氯化钯为(ch3cn)2pdcl2。
优选的,所述步骤(2)中邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物的纯化方法包括以下步骤:将制得的粗产品用乙醚洗涤,真空干燥,制得邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物。
本发明所要解决的技术问题之六在于提供邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物在乙烯聚合中的应用。
本发明通过以下技术手段实现解决上述技术问题的:邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物在乙烯聚合中的应用。
有益效果:邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物可有效催化乙烯齐聚反应,得到高产率的油状低聚物,其结构主要以c10–c24的成分。
优选的,所述邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物用于乙烯聚合的方法包括以下步骤:将邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物与助催化剂以1:100~1000的摩尔比组成复合催化体系,催化乙烯聚合,得到乙烯低聚物。
优选的,所述催化体系中反应温度为20~80℃,反应时间为10~120min,邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物用量为2~10μmol/l。
本发明的优点在于:
(1)本发明制备的邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物,由于邻位二苯甲基的存在而使催化剂金属中心的电子云密度和金属中心周围的空间位阻效应发生变化,在助催化剂氯化二乙基铝的活化下催化乙烯,催化剂催化活性达到1.90×105gofpe(molni)-1h-1,表现出显著的热稳定性,并且提高了聚合活性和分子量,得到低度支化(28个支链/1000c)的半结晶聚乙烯,制得的聚合物具有较好的可塑性,可作为弹性体;
(2)在亚胺氮原子芳环上对位引入了氟原子通强拉电子效应来有效的改变链行走乙烯聚合的行为,相对于对位甲基取代的催化剂,拉电子氟的催化剂进一步提高了催化活性;
(3)本发明制备的邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物催化乙烯的产物为c10–c24的油状低聚物,可以有效催化乙烯齐聚反应,该配合物催化乙烯齐聚反应具有高的催化活性,催化活性达到3.45×105g(molni)-1h-1。
附图说明
图1为本发明阳离子含有邻位二苯甲基取代的亚胺膦镍(ⅱ)和钯(ⅱ)催化乙烯反应过程简图;
图2为本发明实施例2中催化剂ni2催化获得聚乙烯的13cnmr谱;
图3为本发明实施例3中钯催化剂pd1的单晶结构图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所用的试验材料和试剂等,如无特殊说明,均可从商业途径获得。
实施例中未注明具体技术或条件者,均可以按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。
实施例中使用的原料和试剂:所有金属有机反应都在n2保护下进行,溶剂均经干燥除氧处理,二氯甲烷和邻二氯苯(分析纯),用
实施例1
(一)2,6-(ph2ch)2-4-me-c6h2-n=ch-c6h4-2-pph2配体的制备
(1)将4-甲基-2,6-二苯甲基苯胺(0.88g,2.00mmol)和2-二苯基膦苯甲醛(0.58g,2.00mmol)溶解于30ml乙醇中,在搅拌下加入甲酸(0.2ml),在50℃下反应24h后,除去溶剂,得粗产品;
(2)将步骤(1)中制得的粗产品用c2h5oh/ch2cl2(v/v=13:2)混合溶剂重结晶析出固体沉淀,过滤后,真空45℃下干燥得到亮黄色固体粉末1.21g。
其反应式如下:
上述方法制备的产物经核磁共振波谱(nmr)采用美国mercury-400plus核磁共振仪进行测定。
实验结果:本实施例中制得的2,6-(ph2ch)2-4-me-c6h2-n=ch-c6h4-2-pph2配体,其产率为85%,测定结果如下所示:
1hnmr(400mhz,cdcl3,ppm):δ7.35–7.27(m,6h),7.24–7.19(m,6h),7.16–7.09(m,13h),6.91(d,j=7.2hz,9h),6.84–6.80(m,1h),6.59(s,2h),5.31(s,2h),2.13(d,j=6.9hz,6h,-ch3).
13cnmr(100mhz,cdcl3)δ:163.57(n=ch),148.69,144.45,142.92,139.16,138.96,138.45,134.31,134.05,133.89,133.15,131.23,130.12,129.84,129.68,128.57,128.51,127.99,125.93,51.56(-chph2),21.09(-ch3).
31pnmr(162mhz,cdcl3)δ:-10.60ppm.anal.calcd.forc52h42np:c,87.73;h,5.95;n,1.97.found:c,87.75;h,5.97;n,1.95.ft-ir(kbr):1642cm-1(vc=n).
(二)邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物的制备
在氮气保护下,向100ml干燥的反应管中加入上述配体(0.36g,0.50mmol),再加入nibr2(dme)(0.16g,0.50mmol)和二氯甲烷25ml,在室温下搅拌反应12h,过滤悬浮液,母液在真空下除去溶剂,残留物用乙醚(3×15ml)洗涤三次,真空干燥后得到粉末状固体配合物0.43g,命名为ni1,产率为92%。
其反应式如下:
anal.calcdforc52h42br2nnip:c,67.13;h,4.55;n,1.51.found:c,67.15;h,4.56;n,1.53.ft-ir(kbr):1653cm-1(νc=n).
(三)乙烯聚合
(1)将带有磁搅拌子的100ml聚合瓶真空-氮气循环置换三次,在氮气气氛下,加入30ml甲苯溶液,再通入乙烯,使乙烯充分吸收直至饱和,然后用带有刻度的注射器将助催化剂加入反应瓶中,调节乙烯通入量使聚合体系的压力维持在8.0atm,设定温度为20℃并维持反应搅拌10min后,再用注射器加入步骤(二)中制得的镍(ⅱ)配合物的二氯甲烷溶液(2ml,5.0μmol)。在设定聚合温度下,将乙烯聚合反应进行1个小时后,加入5%酸化甲醇溶液(100ml)终止反应,振荡沉淀出聚合产物。过滤沉淀物,用无水甲醇充分洗涤,50℃真空干燥12h,得聚乙烯产品,催化乙烯反应过程如图1所示。
(2)评价聚合性能和分析聚合物结构:称量所得聚合物计算转化率,核磁(nmr)分析聚合物的支化度和微观结构,凝胶渗透色谱(gpc)测分子量和分散度,差热分析(dsc)分析玻璃化温度和熔点。
(3)筛选助催化剂及al/ni摩尔比
在室温下al/ni摩尔比为300,8atm的乙烯压力下,选用本实施例中制得的催化剂ni1,在不同助催化剂(甲基铝氧烷mao,改性的甲基铝氧烷mmao,氯化二乙基铝et2alcl和etalcl2)条件下进行乙烯聚合反应。
实验结果:
(1)不同的助催化剂下进行乙烯聚合结果如表1所示;
表1.在不同的助催化剂下进行乙烯聚合a
其中:a聚合条件为:ni=5μmol;23℃;1h;2mlch2cl2,30ml甲苯;8atm乙烯压力;
b活性单位为105gofpe(molni)-1h-1;
c分子量mn单位kgmol-1,通过gpc检测;
从表1可以看出,在室温下,所用et2alcl作为助催化剂时,催化剂ni1表现出最高的活性和所得最高的分子量。
(2)聚合温度对乙烯聚合的影响:表2为聚合温度为25~75℃时,催化剂ni1在et2alcl的活化下进行乙烯的聚合结果
表2.在不同聚合温度在进行乙烯聚合a
其中:a聚合条件:ni=5μmol;助催化剂et2alcl,[al]/[ni]=500;1h;2mlch2cl2,30ml甲苯;8atm乙烯压力;
b单位105gofpe(molni)-1h-1
c分子量mn单位kgmol-1,通过gpc检测;
d支化度,单位每1000碳包含的支链数,通过1hnmr检测;
e熔点tm通过dsc检测。
从表2可以看出,在50℃下,最高的活性和所得最高的分子量被观察到。得到低度支化度的半结晶聚乙烯,支化度随聚合温度的升高而增大。
实施例2
(一)2,6-(ph2ch)2-4-f-c6h2-n=ch-c6h4-2-pph2配体的制备
(1)将4-氟-2,6-二苯甲基苯胺(0.89g,2.00mmol)和2-二苯基膦苯甲醛(0.58g,2.00mmol)溶解于30ml乙醇中,在搅拌下加入甲酸(0.2ml),在50℃下反应24h后,除去溶剂,得粗产品;
(2)将步骤(1)中制得的粗产品用c2h5oh/ch2cl2(v/v=13:2)混合溶剂重结晶析出固体沉淀,过滤后,干燥得到黄色固体1.30g。
其反应式如下:
上述方法制备的产物经核磁共振波谱(nmr)采用美国mercury-400plus核磁共振仪进行测定。
实验结果:本实施例中制得的2,6-(ph2ch)2-4-f-c6h2-n=ch-c6h4-2-pph2配体,其产率为91%,测定结果如下所示:
1hnmr(400mhz,cdcl3,ppm):δ7.38–7.27(m,6h),7.25–7.20(m,6h),7.18–7.10(m,13h),6.93(d,j=7.2hz,9h),6.86–6.82(m,1h),6.64–6.47(m,2h),5.35(s,2h).
13cnmr(100mhz,cdcl3)δ:163.63(n=ch),148.70,144.46,142.94,139.18,138.97,138.46,134.33,134.06,133.89,133.16,131.24,130.13,129.85,129.69,128.58,128.53,127.99,125.94,51.58(-chph2).
31pnmr(162mhz,cdcl3)δ:-10.63ppm.anal.calcd.forc52h42np:c,87.73;h,5.95;n,1.97.found:c,87.75;h,5.97;n,1.95.ft-ir(kbr):1645cm-1(vc=n).
(二)邻位二苯甲基取代的亚胺膦镍(ⅱ)配合物的制备
在氩气保护下,向100ml干燥的反应管中加入上述配体(0.73g,1.0mmol),再加入nibr2(dme)(0.34g,1.1mmol)和二氯甲烷20ml,在室温下搅拌反应12h,过滤悬浮液,母液在真空下除去溶剂,残留物用乙醚(3×15ml)洗涤三次,真空干燥后得到粉末状固体配合物0.88g,命名为ni2,产率为93%。
其反应式如下:
测定结果如下:anal.calc.forc52h38br2f2n2ni(947.37):c,65.93;h,4.04;n,2.96.found:c,68.58;h,4.51;n,2.76.
(三)乙烯聚合
(1)将带有磁搅拌子的100ml聚合瓶真空-氮气循环置换三次,在氮气气氛下,加入30ml甲苯溶液,再通入乙烯,使乙烯充分吸收直至饱和,然后用带有刻度的注射器将助催化剂氯化二乙基铝(et2alcl)(2.75ml,0.9m,n(al)/n(ni)=500)加入反应瓶中,调节乙烯通入量使聚合体系的压力维持在8.0atm,设定温度为20℃并维持反应搅拌1min后,再用注射器加入上述含镍配合物的二氯甲烷溶液(2ml,5.0μmol)。在设定温度下,将乙烯聚合反应进行1h后,加入5%酸化甲醇溶液(100ml)终止反应,振荡沉淀出聚合产物。过滤沉淀物,用无水甲醇充分洗涤,50℃真空干燥12h,催化乙烯反应过程如图1所示。
实验结果:在25℃聚合温度下,得聚乙烯0.59g,活性为1.17×105gpe/(molnih),重均分子量为2.13×104gmol-1,分子量分布2.48,支化度为28个支链/1000c,熔点126℃。
在50℃聚合温度下,得聚乙烯0.95g,活性为1.90×105gpe/(molnih),重均分子量为1.22×104gmol-1,分子量分布2.79,支化度为39个支链/1000c,熔点121℃。
图2为在25℃,催化剂ni2催化获得聚乙烯的13cnmr谱,从图2可以看出,所得较低支化度聚乙烯的结构主要是甲基支链。
实施例3
(一)邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物的制备
在氮气保护下,向100ml干燥的反应管中加入实施例1中制得的2,6-(ph2ch)2-4-me-c6h2-n=ch-c6h4-2-pph2配体(0.72g,1.0mmol),再加入(mecn)2pdcl2(0.26g,1.0mmol)和二氯甲烷30ml,在室温下搅拌反应24h,过滤悬浮液,母液在真空下除去溶剂,残留物用乙醚(3×15ml)洗涤三次,真空干燥后得到粉末状白色固体配合物0.76g,命名为pd1,产率为89%。
其反应式如下:
测定结果如下:anal.calcdforc52h42cl2nppd:c,70.24;h,4.76;n,1.58.found:c,70.26;h,4.74;n,1.59.ft-ir(kbr):1653cm-1(νc=n).
钯催化剂pd1的单晶结构图如图3所示,图中cl、n、pd、p均已标注,从图3可以看出,催化剂pd1显示出n-芳基邻位上含有两个庞大的2,6-二苯甲基基团,pd中心由n,p和cl以略微扭曲的平面方形模式配位。
(二)乙烯齐聚反应
将带有磁搅拌子的100ml聚合瓶真空-氮气循环置换三次,在氮气气氛下,加入30ml甲苯溶液,再通入乙烯,使乙烯充分吸收直至饱和,然后用带有刻度的注射器将助催化剂氯化二乙基铝(et2alcl)(2.75ml,0.9m,n(al)/n(ni)=500)加入反应瓶中,调节乙烯通入量使聚合体系的压力维持在8.0atm,设定温度为20℃并维持反应搅拌10分钟后,再用注射器加入上述含钯(ii)配合物的二氯甲烷溶液(2ml,10μmol)。在设定温度下,将乙烯齐聚反应进行2小时后,将压力容器放空,用硅胶过滤所得混合物。在真空下获得油状低聚物。称量所得油状产品计算转化率,评价聚合性能,催化乙烯反应过程如图1所示。
实验结果:
(1)在50℃聚合温度下,得油2.14g,活性为1.07×105g/(molpdh);
在75℃聚合温度下,得油6.89g,活性为3.45×105g/(molpdh);
在75℃聚合温度下,得油4.17g,活性为2.35×105g/(molpdh)。
(2)分析聚合物结构:核磁(nmr)分析微观结构,gc和gc-ms分析所含不同产物的含量。
聚合结果揭示:在75℃下,本发明涉及的邻位二苯甲基取代的亚胺膦钯(ⅱ)配合物在助催化剂氯化二乙基铝的活化下催化乙烯齐聚反应。所得的产物为c10–c24的油状低聚物。
表3.pd1-et2alcl催化乙烯齐聚反应所得产品分析a
其中:a聚合条件:pd1=10μmol;助催化剂et2alcl,[al]/[ni]=500;2h;2mlch2cl2,30ml甲苯;8atm乙烯压力;
b通过gc分析各合成物的含量。
对比例1
在实施例1乙烯聚合步骤(1)的聚合条件下,选用2,6-异丙基取代的亚胺膦镍配合物ni3作为催化剂,在25℃聚合温度下,得聚乙烯0.21g,活性为0.42×105gpe/(molnih),重均分子量为0.70×104gmol-1,分子量分布2.96,支化度为57个支链/1000c,熔点107℃。
2,6-异丙基取代的亚胺膦镍配合物ni3和ni1的结构对比如下:
比较催化剂ni3和ni1的数据,聚合结果揭示:本发明涉及的邻位二苯甲基取代的亚胺膦镍镍(ⅱ)配合物,由于邻位二苯甲基的存在而使催化剂金属中心的电子云密度和金属中心周围的空间位阻效应发生变化,此类镍催化剂在助催化剂氯化二乙基铝的活化下催化乙烯,表现出显著的热稳定性,并且提高了聚合活性和分子量,得到较低支化度的半结晶聚乙烯。
对比例2
在实施例1乙烯聚合步骤(1)的相似聚合条件下,选用不对称含有2,6-二苯甲基取代的α-二亚胺镍(ⅱ)的类似物ni4作为催化剂,在25℃聚合温度聚合20分钟下,得聚乙烯3.75g,活性为2.25×106gpe/(molnih),重均分子量为7.1×105gmol-1,分子量分布1.66,支化度为81个支链/1000c,无熔点。在50℃聚合温度聚合20分钟下,得聚乙烯4.17g,活性为2.50×106gpe/(molnih),重均分子量为6.8×104gmol-1,分子量分布1.93,支化度为89个支链/1000c,无熔点。
比较催化剂ni4和ni1的数据,聚合结果揭示:在助催化剂氯化二乙基铝的活化下,二亚胺镍催化剂ni4比亚胺膦镍催化剂ni1催化活性高,得到含支链的高分子量的无定型的聚乙烯(无熔点)。虽然亚胺膦镍催化剂ni1的活性相对低一些,但是所得到了聚乙烯具有较低的支化度,有较高的熔点和结晶度。在烯烃聚合领域,制备具有高性能的聚烯烃材料是一项非常重要的挑战。半结晶高分子聚乙烯具有一定的力学性能,可作为弹性体。
对比例3
在实施例1乙烯聚合步骤(1)的相似聚合条件下,选用对位含芳基取代的α-二亚胺镍配合物作为催化剂,在25℃聚合温度下,得到258-273支化度/1000c的无定型聚乙烯,可塑性差。
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种盐酸替匹嘧啶的晶型及其制备方法与流程
- 一种含有肟菌酯和榄香素的高效杀菌组合物的制造方法与工艺
- 杀虫、杀螨、杀线虫、杀软体动物、杀菌或杀细菌组合物及病害虫的防除方法与制造工艺
- 包含邻近氨基酸端的志贺毒素A亚基效应子区和细胞靶向性免疫球蛋白型结合区的蛋白的制造方法与工艺
- N-酰基亚氨基杂环化合物的制造方法与工艺
- 一种甲氧卡那霉素药物中间体γ‑邻苯二甲酰亚氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种利用光敏氧化由双甘膦合成草甘膦的方法与制造工艺
- N‑羟甲基草甘膦或其酯的合成及其制备草甘膦的方法与制造工艺
- 2-(4-噻唑啉酮-2-亚氨基)噻唑-5-羧酸酯及其制备方法与应用与制造工艺
- 抗流感药物帕拉米韦关键中间体的制备方法与制造工艺