一种高效的酿酒酵母无痕基因敲除方法及其应用与流程
本发明涉及基因工程领域,具体是在酿酒酵母中构建一种高效的无痕基因敲除方法并用该方法构建了一株低产高级醇的酿酒酵母菌株。
背景技术:
酿酒酵母是一种典型的真核细胞模型和重要的模式生物,它的特性包括:生长繁殖快、代谢周期短、易于分离和培养等,这些特性使酿酒酵母更方便于进行基因工程和遗传学研究,称为真核生物中的“大肠杆菌”。随着分子生物学以及基因工程技术的不断发展和不断更新,酿酒酵母的育种方法已经由最初的自然育种发展为现代的定向基因工程育种。基因敲除(geneknockout)是20世纪80年代末发展起来的一种遗传工程技术,是通过适当的方法使机体特定的基因缺失或失活,以达到定点修饰改造染色体上某一基因的目的的一种技术。但是随着公众对食品安全的关注,通过传统遗传学方法构建的菌株,由于残留非本身的外源基因,其生产应用受到限制。因此应用无痕敲除技术敲除机体的特定基因成为目前遗传育种的发展方向。
酵母菌的无痕修饰起初是为了除去筛选标记,以便在单个菌株中进行多基因操作。去除筛选标记主要有两种方法,一种是利用重组酶介导的敲除系统,例如cre/loxp系统,cre重组酶能介导两个loxp位点之间发生特异性重组,把两个位点间的基因删除。通过第一次化学转化,将带有同向两个loxp位点和筛选标记的片段整合到基因组上;然后通过第二次化学转化导入带有编码cre重组酶基因的质粒,利用cre重组酶的表达切除两个位点之间所有序列,从而完成筛选标记的去除。2004年,iwaki等人将该系统应用于酵母中,实现了对酵母的多基因敲除。cre/loxp方法具有很高的效率,但是会在染色体上残留一个外源序列(loxp位点),当进行多基因敲除时候,残留的loxp位点增加了染色体重排的可能。另一种方法则是利用敲除元件中正向重复序列(hisg)之间的同源重组,首先需要构建带有“hisg—ura3—hisg”的质粒,然后使用长引物pcr直接得到敲除元件,通过转化敲除目的基因后,再反向筛选,利用正向重复序列(hisg)之间的同源重组去除筛选标记。2012年,dong等人应用该方法实现了酵母基因的无痕敲除,但是该方法在诱导第二步同源重组的几率是随机的,产生的转化子不只一种,使得最终得到理想转化子的几率变低。2013年初,一种全新的人工核酸内切酶clusteredregularlyinterspacedshortpalindromicrepeats(crispr)/crispr-associated(cas))出现,它主要是基于细菌的一种获得性免疫系统改造而成,其特点是制作简单、成本低、作用高效。2014年,bao等首次利用crispr-cas系统对酿酒酵母实现多基因一步敲除。但是该系统中设计的向导rna(sgrna)在指导cas蛋白与基因组进行识别时会与非靶点dna序列错配,引入非预期的基因突变,即脱靶效应,严重制约了该技术的广泛应用。总之,本领域仍需要一种高效的无痕基因敲除方法。
高级醇是酿酒酵母酒精发酵过程中的重要代谢产物,也是白酒、葡萄酒等酒精饮料中主要的风味物质,其含量的多少及各种醇之间的比例对酒的风味有着重要的影响,适量的高级醇和协调的组分比可以赋予酒特殊的香味,同时衬托出酯的香气,使酒的口感协调、柔和。而高级醇含量过高会使酒产生异杂味,影响酒的风味和品质,并且高级醇在人体内的氧化速度比乙醇慢,其对人体的毒害作用远远高于乙醇。因此,控制高级醇含量是现代酿造过程中一项重要的指标。在酿酒酵母酒精发酵过程中,有两条高级醇代谢途径,分别为糖代谢合成途径(harris途径)和氨基酸分解代谢途径(ehrlich途径)。在氨基酸分解代谢途径中,酿酒酵母支链氨基酸分解代谢的第一步是转氨作用。据研究报道表明,bat2基因编码的支链氨基酸转氨酶在高级醇特别是异丁醇和异戊醇的生成过程中起着非常重要的作用。因此,为了降低发酵过程中高级醇的生成量,敲除对高级醇有重要影响的氨基酸转氨酶编码基因bat2将是一条有效的途径。
因此,本发明将以氨基酸转氨酶编码基因bat2为靶基因在酿酒酵母中构建一种高效的无痕敲除方法,该方法可广泛应用于酵母及其他微生物的基因改造,为直接在工业菌株中进行基因敲除提供了有益的参考。构建的低产高级醇突变菌株可安全用于工业生产,满足酿酒酵母应用相关领域对酵母的要求。
技术实现要素:
本发明解决的技术问题是提供一种高效的酿酒酵母无痕基因敲除的方法,并用该方法构建了一株低产高级醇的酿酒酵母菌株。
为解决上述技术问题,本发明采用如下技术方案:
一种高效的酿酒酵母无痕基因敲除方法,以酿酒酵母ay15的单倍体α5为出发菌株,bat2基因为靶基因,实现对基因bat2的无痕敲除。
本发明所构建的低产高级醇酵母菌株是对出发菌株酿酒酵母(saccharomycescerevisiae)cicc32315单倍体α5中的完整氨基酸转氨酶编码基因bat2进行无痕敲除来实现。具体为酿酒酵母hα5(500)δh。
所述bat2基因其geneid为:853613,核苷酸序列如表所示。
上述基因无痕敲除可以用以下方法实现。各步骤所涉及具体操作方法参考现有文献报道,如josephsambrook等,《分子克隆实验指南》第二版,科学出版社,1995。
用pcr方法扩增出herp1.0片段,将该片段克隆至yep352质粒,构建yherp1.0质粒;用pcr方法分别扩增敲除靶基因bat2上游和下游长度为500bp和683bp的同源序列片段,然后通过融合pcr获得无缝融合片段;将该片段克隆至质粒yherp1.0上,获得重组质粒yherp1.0(500);用pcr方法分别扩增敲除靶基因上游597bp及yherp1.0(500)上的片段;通过醋酸锂转化法将这两个片段同时导入酿酒酵母中,经过ypgly+af筛选培养基进行第一步整合的筛选,获得含抗性基因的突变株;用半乳糖诱导培养基30℃,180rpm培养24h,稀释100倍后涂布于含有5-氟-2’-脱氧尿苷的合成培养基平板上,30℃培养36h,获得第二步整合重组的酿酒酵母菌株。
所述酿酒酵母在生长性能与发酵性能不受影响情况下,在第二次整合重组敲除筛选标记过程的概率为6.86×10-4。
本发明所述酿酒酵母菌株(hα5(500)δh)可以用于白酒生产中。
本发明同时提供了对于诱导培养基的优化,探究了诱导培养基中半乳糖的含量对其第二步整合重组概率的影响如表1,操作过程如下:
1、诱导培养基中半乳糖的含量共设制0.1g/100ml,0.5g/100ml,1g/100ml,2g/100ml,3g/100ml,4g/100ml,5g/100ml八个梯度,蛋白胨2g/100ml,酵母浸粉1g/100ml。
2、将hα5(500)突变株用半乳糖培养基进行诱导培养24h。稀释100倍后涂布含有5-氟-2’-脱氧尿苷的合成培养基平板。
3、表1第二步整合重组的概率
本发明的积极效果如下:
1、本发明提供一种高效的酿酒酵母无痕基因敲除的方法,克服了传统基因敲除中筛选标记残留、不便进行多基因敲除的困难。同时该发明不仅可以用于研究酵母基因的功能和代谢机制,而且由于所获得的突变株不残留任何外源基因,可以安全的用于工业生产。
2、本发明提供的低产高级醇酿酒酵母在保持良好发酵性能的前提下,完全敲除氨基酸转氨酶编码基因bat2,通过玉米浓醪白酒发酵实验,改造菌株的正丙醇、异丁醇和异戊醇含量有显著的降低,达到了低产高级醇的目的,为酿造出风味优良且更有利于健康的酒精饮料奠定了理论基础。
附图说明:
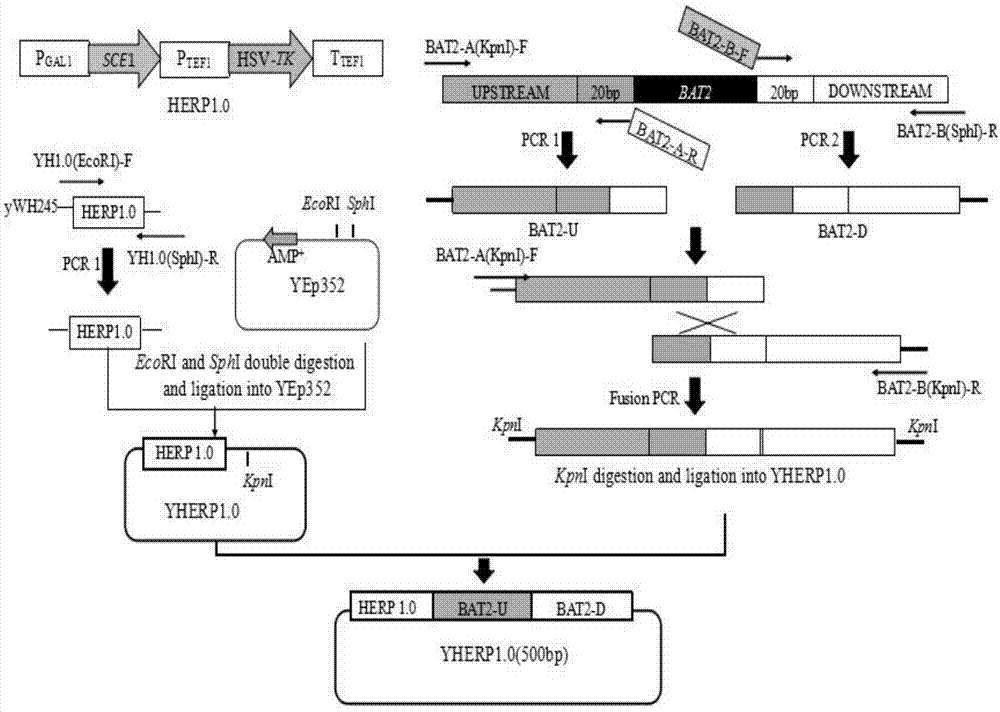
图1为重组质粒yherp1.0(500)的构建流程示意图。
图2为重组质粒yherp1.0与yherp1.0(500)的构建与验证电泳图。
图3为无痕敲除过程中的两步整合重组流程示意图。
图4为发生第一步整合重组的菌株hα5(500)的电泳验证图。
图5为发生第二步整合重组的菌株hα5(500)δh电泳验证图。
图6为整合重组菌株hα5(500)δh靶基因位置的部分测序比对图,其中图6a为敲除前后序列对比,图6b为敲除后靶位置40bp的测序结果。
图7为hα5(500)δh与α5的生长曲线。
具体实施方式:
下面通过具体的实施方案叙述本发明。除非特别说明,本实验中所用的技术手段均为领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。
实施例1
无痕敲除氨基酸转氨酶编码基因bat2酿酒酵母的构建
本实例所用的出发菌株为cicc32315的单倍体α5。(所述片段herp1.0来源见williamg.alexander,drewt.doeringandchristoddhittinger,high-efficiencygenomeeditingandallelereplacementinprototrophicandwildstrainsofsaccharomyces.genetics,vol.198,859–866november2014)。所述大肠杆菌dh5α购自takara公司。所述ypd培养基为通用的完全培养基;所述的筛选培养基ypgly+af成分为5%甘油、2%蛋白胨、1%酵母浸粉、200mg/ml氨甲基叶酸、5mg/ml磺胺、5μg/ml胸腺嘧啶,50μg/ml次黄嘌呤;所述的酵母合成培养基(sc)成分为2%葡萄糖、0.17%ynb、0.5%硫酸铵、完全氨基酸混合物溶液,固体培养基含2%进口琼脂粉。
根据genebank中的酵母基因组数据和yep352质粒序列,设计了下述实施例中各引物。
表2本实施例中所用到的引物
注:下划线加斜体表示酶切位点,斜体表示融合pcr所用引物的重叠序列,下划线表示添加的酶切位点。
表3本实施例中所用的pcr扩增体系
表4本实施例中所用的融合pcr体系
(1)质粒yherp1.0的构建
质粒的构建流程如图1所示;
首先,使用solarbio公司的酵母基因组提取试剂盒,提取ywh245的基因组。利用引物yh1.0(ecori)-f和yh1.0(sphi)-r,以ywh245的基因组为模板,pcr扩增菌株ywh245中的herp1.0片段,经过试剂盒回收;用ecori和sphi内切酶对质粒yep352进行双酶切,切胶回收酶切质粒;用vazyme的clone
(2)重组质粒yherp1.0(500)的构建
重组质粒yherp1.0(500)的构建如图1所示
首先用solarbio公司的酵母基因组提取试剂盒,提取cicc32315的单倍体α5的基因组。利用引物bat2-a(kpni)-f和bat2-a-r扩增bat2基因上游500bp;同时用引物bat2-b-f和bat2-b(kpni)-r扩增bat2基因下游683bp,分别命名bat2-u和bat2-d。然后以bat2-u和bat2-d的混合物为模板,加入引物bat2-a(kpni)-f和bat2-b(kpni)-r进行融合pcr,获得无缝融合片段,命名为bat2-ud。经过切胶回收(试剂盒)后,用kpni进行酶切,亚克隆到质粒yherp1.0的相应酶切位点中,并用引物ahb-b-u与bat2-b(kpni)-r验证单酶切连接的正反,重组质粒yherp1.0(500)构建成功。
图2为重组质粒yhherp1.0与yhherp1.0(500)的构建与验证电泳图:泳道1为herp1.0(3159bp)电泳结果;泳道2为ecori和sphi酶切yep352质粒(5100bp)电泳结果;泳道3为yherp1.0质粒电泳结果:泳道4为yep352质粒电泳结果;泳道5为kpni酶切yherp1.0质粒(8300bp)电泳结果;泳道6为bat2-u1d片段(1183bp)电泳结果;泳道7为yherp1.0(500)质粒电泳结果;泳道8为yherp1.0(500)单酶切连接验证正反(1260bp)电泳结果;泳道9为ecori酶切yherp1.0(500)质粒(3800bp和5700bp)电泳结果;泳道m为15000dnaladdermaker。
上述融合pcr法为本领域公知的采用具有互补末端的引物,形成具有重叠序列pcr产物,通过pcr产物重叠序列延伸,从而将任意dna片段连接起来方法,此技术不需要内切酶的消化和连接酶的处理,就可实现dna的体外连接。
(3)酿酒酵母中氨基酸转氨酶编码基因(bat2)的敲除
用引物bat2-a-f和bat2-a-rf扩增bat2左侧597bp,得到635bp长度片段,命名bat2-a—sf;同时用引物bat2-b-fr和bat2-b(kpni)-r在yherp1.0(500)上扩增得到4700bp左右长度的片段,命名为rs—herp1.0—bat2-ud;用醋酸锂转化法将片段bat2-a—sf与rs—herp1.0—bat2-ud导入酿酒酵母菌株α5中,经过两步整合重组后,得到无痕敲除bat2的酿酒酵母,两步重组过程如图3。
第一步整合重组的发生,是由于导入的两个片段与酵母基因组的同源部分发生整合,而两个片段之间存在同源部分也会发生整合,从而使整个片段整合到α5的基因组。转化后的悬浮液,涂布于ypgly+af筛选培养基平板上,30℃培养7d,获得发生第一步整个重组的酵母菌株hα5(500)。随机挑选所得的单菌落,采用ahb-a-u和ahb-a-d为整合位点上游验证引物,ahb-b-u和ahb-b-d为整合位点下游验证引物,进行菌落pcr筛选。图4为发生第一步整合重组的菌株hα5(500)的电泳验证图:泳道m为dl5000dnaladdermaker;泳道1,2分别使用上游验证引物pcr验证结果,泳道1模板为hα5(500),泳道2模板为α5;泳道3,4分别使用下游验证引物pcr验证结果,泳道3模板为hα5(500),泳道4模板为α5。
经过筛选和鉴定获得的重组酿酒酵母菌株hα5(500),取一环接到5ml半乳糖诱导培养基中,30℃,180rpm培养24h,稀释100倍涂布于含有5-氟-2’-脱氧尿苷的合成培养基平板上,30℃培养36h,获得第二步整合重组的酿酒酵母菌株。随机挑选所得到的单菌落,采用ahb-a-u和ahb-b-d引物进行菌落pcr筛选,经验证第二整合重组的概率可以达到6.86×10-4,可以快速、高效的做到筛选标记的回收,便于再次利用,并且达到无痕敲除的目的。图5为发生第二步整合重组的菌株hα5(500)δh的电泳验证图:泳道m为dl5000laddermaker;泳道1为使用引物ahb-a-u和ahb-b-d进行pcr的验证结果,泳道1模板为hα5(500)δh。
为了进一步验证敲除靶位置的序列情况,提取无痕敲除菌株hα5(500)δh的基因组,使用引物ahb-a-u和ahb-b-d进行pcr扩增,获得2605bp的片段送于华大基因公司测序,测序结果如图6。图6a为敲除前后序列对比,图6b为敲除后靶位置40bp的测序结果,其他结果未给出,可以确定无痕敲除基因bat2酿酒酵母菌株构建成功。
实施例2
无痕敲除bat2酿酒酵母菌株hα5(500)δh与α5生长曲线的测定
挑取hα5(500)δh与α5的单菌落接种于50mlyepd液体培养基中,30℃,180rpm振荡培养24h,取扩大培养的菌液按照1:100的接种量接种于3瓶50mlyepd培养基中,同样条件下振荡培养,并分别于培养后0h、1h、2h、3h、4h、5h、6h、7h、8h、9h、10h、11h、12h、13h、14h、15h取菌体培养悬液0.5ml,10000rpm离心1min,然后用去离子水重悬菌泥。以去离子水校正7200型可见分光光度计的零点,在波长600nm处比色测定菌液od600值。以各时间点菌液的吸光光度值(od600)为纵坐标,以培养时间为横坐标,绘制出各菌株的生长曲线,生长曲线如图7,从生长曲线可以看出hα5(500)δh与出发菌株α5生长性能并没有差别。
实施例3
无痕敲除bat2酿酒酵母菌株hα5(500)δh与α5的玉米浓醪发酵实验
将重组菌株hα5(500)δh和出发菌株α5分别同时进行玉米浓醪发酵实验,发酵工艺路线图:
玉米粉浸泡→液化→糖化→加菌发酵→称量失重→蒸馏酒精→测定发酵指标
工艺条件:
浸泡条件,60-70℃,浸渍20min;液化条件:85-90℃,加入耐高温α-淀粉酶,液化90min;糖化条件,55-60℃,加入糖化酶,糖化20min。
配料:玉米粉60g,水130ml,耐高温α-淀粉酶2×104u/ml,30μl,糖化酶1×105u/ml,90μl,7.5×102u/ml酸性蛋白酶1.2ml:营养盐1ml(mgso4150g/l、kh2po475g/l、尿素81g/l,过滤,4℃保存);
挑取一环酵母细胞,接入装有5ml一级种子培养基的试管中,30℃静置培养,按10%接种量接种到装有45ml二级种子培养基的三角瓶中,30℃静置培养16h至对数期的后期,按10%接种量接种到玉米浓醪发酵培养基,30℃静置发酵。每隔12h称重一次,当两次失重小于1g,发酵结束。发酵结束后取浓醪100ml,水100ml,蒸出100ml酒样。测定失重(即co2累积排放量)、酒精度和残留还原糖等发酵性能指标,结果如表3,重组菌株发酵后的酒精含量和残糖含量与出发菌株相比没有明显差别,本例中的基因敲除不会对菌株基本发酵性能有不利影响。
表3重组菌株hα5(500)δh和出发菌株α5的发酵性能测定
注:所示数据为三个平行实验结果的平均值。
气相色谱仪:agilent7890c;色谱柱:白酒专用柱,at.lzp-930,230℃,50m×320μm×1μm;检测器:fid检测器,检测器温度:200℃;载气:高纯氮,流速5ml/min;检测条件:程序升温,50℃保持8min,5℃/min升到120℃,保持8min;进样温度:200℃;进样量:1.0μl;分流方式:分流,分流比10:1;结果如表4.结果显示,重组菌株hα5(500)δh的高级醇中异丁醇含量达到42.24mg/l,比出发菌株α5降低了47.85%;异戊醇含量达到123.96mg/l,比出发菌株α5下降了23.14%。
表4重组菌株hα5(500)δh和出发菌株α5的发酵指标(单位mg/ml)
注:所示数据为三个平行试验结果的平均值。
序列表
<110>天津科技大学
<120>一种高效的酿酒酵母无痕基因敲除方法及其应用
<160>14
<170>siposequencelisting1.0
<210>1
<211>36
<212>dna
<213>引物yh1.0(ecori-funknown)
<400>1
gaccatgattacgaattctggatggacgcaaagaag36
<210>2
<211>45
<212>dna
<213>引物yh1.0(sphi-runknown)
<400>2
gtgccaagcttgcatgcggggtaccattaagggttctcgagagct45
<210>3
<211>28
<212>dna
<213>引物bat2-a(kpni-funknown)
<400>3
ggggtacccgctcctttccaaacatctt28
<210>4
<211>40
<212>dna
<213>引物bat2-a-r(unknown)
<400>4
ccacgagttttaagaacgatagtatcgctattgctacgta40
<210>5
<211>40
<212>dna
<213>引物bat2-b-f(unknown)
<400>5
aagatgtttggaaaggagcggagctcgttttcgacactgg40
<210>6
<211>26
<212>dna
<213>引物bat2-b(kpni-runknown)
<400>6
ggggtacctcaatcggcacattcata26
<210>7
<211>18
<212>dna
<213>引物bat2-a-f(unknown)
<400>7
gctccctccaactactct18
<210>8
<211>56
<212>dna
<213>引物bat2-a-rf(unknown)
<400>8
cttctttgcgtccatccaattaccctgttatccctaatcgttcttaaaactcgtgg56
<210>9
<211>56
<212>dna
<213>引物bat2-b-fr(unknown)
<400>9
ccacgagttttaagaacgattagggataacagggtaattggatggacgcaaagaag56
<210>10
<211>18
<212>dna
<213>引物ahb-a-u(unknown)
<400>10
ttcactgggaccctttca18
<210>11
<211>22
<212>dna
<213>引物ahb-a-d(unknown)
<400>11
gcttctaatccgtacttcaata22
<210>12
<211>18
<212>dna
<213>引物ahb-b-u(unknown)
<400>12
atgcgtcaatcgtatgtg18
<210>13
<211>18
<212>dna
<213>引物ahb-b-d(unknown)
<400>13
tgaaatccttgtccagct18
<210>14
<211>1131
<212>dna
<213>酿酒酵母(saccharomycescerevisiae)
<400>14
atgaccttggcacccctagacgcctccaaagttaagataactaccacacaacatgcatct60
aagccaaaaccgaacagtgagttagtgtttggcaagagcttcacggaccacatgttaact120
gcggaatggacagctgaaaaagggtggggtaccccagagattaaaccttatcaaaatctg180
tctttagacccttccgcggtggttttccattatgcttttgagctattcgaagggatgaag240
gcttacagaacggtggacaacaaaattacaatgtttcgtccagatatgaatatgaagcgc300
atgaataagtctgctcagagaatctgtttgccaacgttcgacccagaagagttgattacc360
ctaattgggaaactgatccagcaagataagtgcttagttcctgaaggaaaaggttactct420
ttatatatcaggcctacattaatcggcactacggccggtttaggggtttccacgcctgat480
agagccttgctatatgtcatttgctgccctgtgggtccttattacaaaactggatttaag540
gcggtcagactggaagccactgattatgccacaagagcttggccaggaggctgtggtgac600
aagaaactaggtgcaaactacgccccctgcgtcctgccacaattgcaagctgcttcaagg660
ggttaccaacaaaatttatggctatttggtccaaataacaacattactgaagtcggcacc720
atgaatgcttttttcgtgtttaaagatagtaaaacgggcaagaaggaactagttactgct780
ccactagacggtaccattttggaaggtgttactagggattccattttaaatcttgctaaa840
gaaagactcgaaccaagtgaatggaccattagtgaacgctacttcactataggcgaagtt900
actgagagatccaagaacggtgaactacttgaagcctttggttctggtactgctgcgatt960
gtttctcccattaaggaaatcggctggaaaggcgaacaaattaatattccgttgttgccc1020
ggcgaacaaaccggtccattggccaaagaagttgcacaatggattaatggaatccaatat1080
ggcgagactgagcatggcaattggtcaagggttgttactgatttgaactga1131
- 还没有人留言评论。精彩留言会获得点赞!