HPV全长基因组质控品的制备方法、扩增引物及检测试剂与流程
本发明涉及基因检测技术,特别涉及一种hpv全长基因组质控品的制备方法、扩增引物及检测试剂。
背景技术:
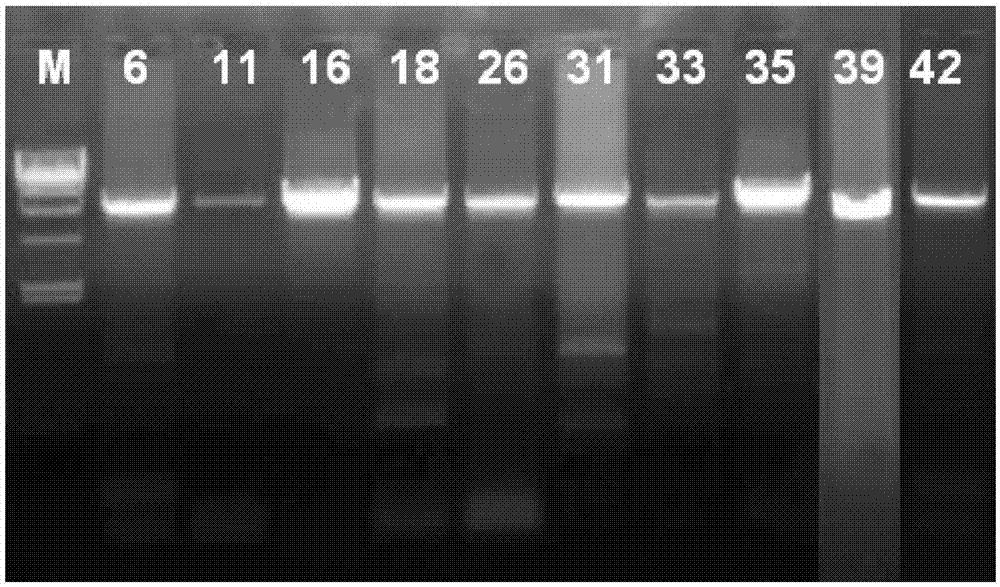
人乳头瘤病毒(humanpapillomavirus,hpv)是一组无包膜的小dna病毒,属乳头瘤病毒科。hpv病毒的形态特征通常为直径52-55nm的正二十面体。其基因组dna长度约8000bp,基因组从功能上分为3个部分:包括早期转录区(编码e1、e2、e4、e5、e6、e7等早期蛋白,参与病毒的复制、转录、翻译调控和转化等功能)、晚期转录区(编码主要衣壳蛋白l1和次要衣壳蛋白l2,用于病毒组装;其中l1约占衣壳蛋白的80%,高度保守,l2含量较少,变异较多)和长控制区(longcontrolregion,lcr,含有hpv基因组dna的复制起点和hpv表达所需的调控元件,调控病毒的转录与复制)。目前已有hpv基因型别100余个,近33%的基因型与生殖道损伤有关,临床上根据hpv的致癌能力分为低危型与高危型hpv。低危型hpv感染主要引起尖锐湿疣,而高危型hpv持续感染则已被确认为导致宫颈病变的主要原因。综合文献报道,高危型hpv包括hpv16、18、26、31、33、35、39、45、51、52、53、56、58、59、66、68、73、82,共18种hpv,常见低危型包括hpv6、11等。由于hpv不能在传统的细胞培养液中培养,其它经典的直接病毒学诊断技术,例如电子显微镜和免疫组织化学,对于hpv的常规检测来说,敏感性和特异性不佳,因此,所有目前用于诊断的hpv检测试剂主要依赖于病毒核酸检测。目前临床常见的hpv检测方法有pcr-反向点杂交法、pcr-荧光探针法、杂交捕获法、流式荧光杂交法、表面等离子谐振法、pcr-质谱法等。常见的靶序列有l1、e6、e7及e1等区域,部分试剂盒的靶序列为整个hpv基因组。hpv分型和检测试剂种类较多,具有不同的原理和特点,分析敏感性、特异性等性能指标也各有差异。国家和政府应当建立相应的与国际标物溯源的标准物质、样本盘或者质控品,对已有的和不断产生的新的试剂进行分析敏感性、临床敏感性、重复性等性能指标进行评价。也应当开展实验室间的室间质量评价,对实验室hpv检测或者基因分型的结果进行比对,以提高实验室的检测水平。英国国家生物学标准品和质控品研究所研究建立了hpv16和18国际标准物质,世界各国均可依据该国际标准物质进行溯源得到其国家标准物质,相应的hpv基因分型试剂厂家亦可依据该国际标准物质或据其溯源的国家标准物质,建立具有统一且可比量值单位的企业参考品,并用于每批试剂的量值溯源和批间质检,从而实现不同试剂方法间的检测结果的可比性。国际标准物质采用重组hpv全基因组质粒和人宫颈细胞系提取的hpv阴性的人基因组dna混合物制备得到。国际标准物质价格昂贵,数量较少,不易购买,且仅有hpv16和18这2个型别,不能覆盖其他常见的hpv型别,不能满足我国试剂标准化、实验室对试剂方法性能评价、室内质量控制和室间质量评价的需要。由中国食品药品检定研究院研制体外诊断试剂国家标准物质人乳头瘤病毒全基因组分型参考品(批号:360003-201101),用于以非l1区域为靶序列的人乳头瘤病毒核酸(基因分型)检测试剂盒(包括以非l1区域如e6、e7、e1区域和全基因组为靶序列)质量控制和评价。该参考品由20种不同型别阳性参考品和阴性参考品n1-n5组成。采用分光光度计测量20种不同hpv基因型的hpv全基因组重组质粒浓度,用1ng/μl人基因组te溶液,将每个型别的hpv重组质粒稀释到106copies/ml左右。包含hpv6、11、16、18、26、31、33、35、45、56、58、59、61、66、67、69、71、73、81、82共20种。上述提及的国家标准物质价格较贵,产量较小,所含基因型未覆盖常见的hpv39、52、68等型别,不能满足我国试剂标准化、实验室对试剂方法性能评价、室内质量控制和室间质量评价的需要。技术实现要素:本发明所要解决的技术问题是:提供一种用于扩增hpv全长基因组的引物;同时还提供一种用于数字pcr检测hpv全长基因组的试剂;在此基础上还提供一种hpv全长基因组质控品的制备方法;可以覆盖包含hpv39、52、68等的23种hpv基因型别,并且满足我国试剂标准化需求。为了解决上述技术问题,本发明采用的技术方案为:一种用于扩增hpv全长基因组的引物,包括用于扩增hpv16型、hpv18型、hpv26型、hpv31型、hpv33型、hpv35型、hpv39型、hpv45型、hpv51型、hpv52型、hpv53型、hpv56型、hpv58型、hpv59型、hpv66型、hpv68型、hpv73型、hpv82型、hpv6型、hpv11型、hpv42型、hpv43型和hpv81型全基因组的扩增引物,其核苷酸序列如seqidno.1-46所示。根据上述,采用上述核苷酸序列如seqidno.1-46所示的引物,可扩增出覆盖包含hpv39、52、68等的23种基因型别的hpv全长基因组。一种用于数字pcr检测hpv全长基因组的试剂,包括用于检测hpv16型、hpv18型、hpv26型、hpv31型、hpv33型、hpv35型、hpv39型、hpv45型、hpv51型、hpv52型、hpv53型、hpv56型、hpv58型、hpv59型、hpv66型、hpv68型、hpv73型、hpv82型、hpv6型、hpv11型、hpv42型、hpv43型和hpv81型的数字pcr上游引物、下游引物和探针,其核苷酸序列依次如seqidno.47-48、seqidno.49-50和seqidno.51所示。根据上述,采用上述核苷酸序列如seqidno.47-48、seqidno.49-50和seqidno.51所示的引物和探针进行数字pcr,可用于检测hpv23种基因型别。一种hpv全长基因组质控品的制备方法,包括以下步骤:(1)获得hpv的dna模版;(2)采用权利要求1所述的用于扩增hpv全长基因组的引物中的扩增引物进行pcr扩增,获得包含hpv全长基因组的目的dna片段,将获得的目的dna片段进行纯化;(3)采用hdcloning连接技术将目的dna片段与载体连接,构建获得质粒;(4)将质粒导入至感受态细胞中,然后提取质粒并验证质粒与hpv各基因型别的序列是否匹配;(5)采用人基因组稀释质粒,获得所述hpv全长基因组质控品。本发明的有益效果在于:(1)利用高保真dna聚合酶的特性,扩增出hpv全基因组dna片段,建立了一种可将8000bp的片段整体高保真高效率扩增的方法;(2)利用hdcloning酶的连接特性,将目的片段无缝、定向的克隆至载体的目的位置,避免连接效率低下的问题;(3)利用数字pcr绝对定量技术,确定了质粒dna的浓度,解决了质控品浓度不确定的问题;(4)成功地得到无生物传染危险性而又可模拟临床样本、可大量生产、浓度可控、稳定保存的hpv基因检测质控品,具有较好的适用性。附图说明图1为本发明实施例的hpv全长基因组质控品的制备方法中样本dna扩增后的pcr产物进行电泳的电泳结果图;图2为本发明实施例的hpv全长基因组质控品的制备方法中样本dna扩增后的pcr产物进行电泳的另一电泳结果图;图3为本发明实施例的hpv全长基因组质控品的制备方法中tvector的结构示意图(含克隆位点);图4为本发明实施例的hpv全长基因组质控品的制备方法中样本选择荧光图谱;图5为本发明实施例的hpv全长基因组质控品的制备方法中标准曲线公式图;图6为本发明实施例的hpv全长基因组质控品的制备方法中稳定性研究起始ct值图;图7为本发明实施例的hpv全长基因组质控品的制备方法中稳定性研究ct值变化图;图8为本发明实施例的hpv全长基因组质控品的制备方法中数字pcr绝对定量法对23种质控品的定值结果图。具体实施方式为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式并配合附图予以说明。本发明最关键的构思在于:将23个型的人乳头瘤病毒全基因组dna克隆成质粒dna,从而实现对hpv基因检测试剂盒的质量控制和评价。由于目前现有的hpv标准物质价格较贵,产量较小,所含基因型未覆盖常见的型别,不能满足我国试剂标准化、实验室对试剂方法性能评价、室内质量控制和室间质量评价的需要。本发明统计了已获得中国食品药品监督管理局认证的63个hpv检测试剂所覆盖的基因型,选择80%以上的试剂可检测的型别,包括18种高危型:hpv16、18、26、31、33、35、39、45、51、52、53、56、58、59、66、68、73、82;5种低危型:hpv6、11、42、43、81。本专利使用到的主要技术有:1、高保真长片段扩增技术:高保真dnapolymerase改良型pcr酶,具有α型dna聚合酶特有的高保真性和优良的扩增性,可以有效抑制非特异性扩增。添加了延伸因子和新开发的特异性促进因子,可以对宽广的模板量进行快速、高特异性扩增。对于常规dna聚合酶难以扩增的高gc含量或高at含量序列,也可以进行有效扩增。此外,反应缓冲液中含有pcr反应的阻害物吸收成分和扩增增强因子,对粗提样品也可以进行高效pcr扩增。添加了常温下能抑制dna聚合酶活性和3’-5’核酸外切酶活性的单克隆抗体,适用于hotstartpcr。2、hdcloning酶克隆技术:hdcloning酶是一种简便高效的克隆酶,能够将单个或者多个dna片段快速定向克隆到任意载体中。hdcloning连接技术是一个能够将单个或者多个dna片段快速定向克隆到任意载体中的克隆技术。hdcloning酶通过识别dna片段和线性化载体末端15bp同源序列,将dna片段和线性化载体高效精确地融合在一起。15bp同源序列是通过扩增目的片段所设计的引物来获得的。尤其表现在长片段、短核苷酸片段和多重片段克隆上,其连接效果显著,成功率高,无需进行限制性酶切处理、磷酸化处理或者连接反应,最终的重组载体不附加任何冗余或者不需要的碱基序列。3、数字pcr绝对定量技术:数字pcr一般包括两部分内容,即pcr扩增和荧光信号分析。在pcr扩增阶段,数字pcr先将样品稀释到单分子水平,再平均分配到几十至几万个单元中进行反应。与qpcr对每个循环进行实时荧光测定的方法不同,数字pcr是在扩增结束后对每个反应单元的荧光信号进行采集,有荧光信号记为1,无荧光信号记为0,有荧光信号的反应单元中至少包含一个拷贝的。理论上,在样品中目标dna浓度极低的情况下,有荧光信号的反应单元数目等于目标dna分子的拷贝数。但是,通常每反应单元中可能包含两个或两个以上的目标分子,就需要使用泊松概率分布函数(poissondistribution)进行计算,根据反应单元总数、有荧光信号的单元数以及样品的稀释系数,就可以得到样本的最初拷贝数(浓度)。本发明要解决的技术关键问题包含以下:1、各型别特异性引物的设计和扩增。引物设计要保证各型别引物扩增时不能出现非特异扩增,且普通的taq酶扩增时容易产生未知突变,失去原有的保真性,需调整好引物浓度及使用高保真的酶;由于hpv全基因组长度约8000bp,扩增程序需要据此研究延长延伸时间,增加循环数。2、pcr产物与载体连接及导入。由于pcr产物长度较大,如使用普通的t/a克隆连接方式,可能会导致连接效率低下,连接不稳定,故需要研究连接酶、连接体系、连接时间,且连接后形成的质粒dna约10000bp,导入感受态细胞的难度颇高,需研究解决转化效率不高、鉴别假阳性菌等问题。3、质粒dna的浓度确定问题。由于提取后的质粒原液浓度测量方式不同,可能导致质控品中目的dna的浓度无法确定,如使用紫外分光光度计测量dna浓度的方法,存在光度准确度±0.2%的误差,如使用实时荧光定量pcr的方法,则存在荧光阈值(threshold)设定偏差、需要建立标准曲线等问题。故需要利用数字pcr定量技术,设计荧光引物、探针,优化反应体系,确定稀释倍数,解决浓度无法确定的问题。本发明应用pcr-hdcloning连接原理实现hpv全基因组质粒制备,叙述如下:选取感染了单一型hpv的宫颈脱落细胞,通过pcr-反向点杂交法确定hpv型别,通过荧光pcr测定hpvdna浓度,选择ct值在25以下的高浓度样本作为dna模版,以提高扩增成功率。统计已经发表的hpv基因序列,找出各区域的起始位置及终止位置,然后根据hpv基因序列特点,针对23个型的基因序列,设计了特异的23对pcr引物;同时,由于hdcloning功能酶的特殊要求,需要在引物的5’端增加与载体序列同源的15bp片段。hpv23个型全基因组扩增引物序列如表1所示:表1(3)由于hpv全基因组长度约8000bp,扩增程序据此延长延伸时间至300s,同时增加循环数至60个;pcr扩增产物经过琼脂糖凝胶电泳仪电泳,根据是否获得了单一的dna片段及其片段长短来确定扩增是否成功;将已知浓度的或者梯度浓度的dna与pcr产物同时进行凝胶电泳,通过比较对pcr产物进行定量,从而评估pcr产物浓度。将目的片段进行切胶回收,得到纯化后的dna片段;优化后的pcr反应体系如表2所示,扩增程序如表3所示:表2基因组dna2ul2×pcrbuffer(mg2+,dntpplus)25uldnapolymerase(1.25units/ul)1ulprimer*f(10um)1ulprimer*r(10um)1uldh2o20ultotal50ul表3退火温度和退火时间对pcr扩增效率和特异性扩增影响较大,上述条件优化结果显示退火温度偏低会有非特异性扩增信号;温度偏高扩增效率偏低,灵敏度下降。(4)利用hdcloning功能酶的特点,将dna片段和载体连接,构成hpv全基因组质粒dna,将反应体系置于50℃孵育15min,然后置于冰上;建立最优的hdcloning克隆反应体系,具体参见下表4:表45xhdenzymepremix2μllinearizedvectort2μlpurifiedpcrfragment6μltotal10ul(5)将质粒导入至感受态细胞中:涂布在一个包含氨苄抗生素的lb平板上,氨苄抗生素浓度为100μg/ml,37℃过夜培养,次日挑取单克隆,通过酶切处理或者pcr筛选确定插入片段成功与否;(6)提取质粒,测序验证序列是否匹配,得到的结果如表5,即为23种基因型的序列比对情况汇总表。表5(7)用人基因组(0.5至2ng/μl)稀释质粒,并分装成质控品盘。(8)使用数字pcr反应体系及其分析系统,确定各质控品中质粒dna的浓度。hpv23个型数字pcr引物及探针序列如表6所示,探针p的5’端标记报告荧光基团fam,3’端标记mgb。优化后的数字pcr反应体系如表7所示,扩增程序如表8所示。表6表7各质控品2ul2xddpcrmix(mg2+,dntpplus,taq,rox,buffer)10ulprimer*f(10um)0.2ulprimer*r(10um)0.2ulprobe*p(10um)0.2uldh2o7.4ultotal20ul表8对于人乳头瘤病毒基因检测,hpvdna检测是目前常见的诊断方法,hpv分型和检测试剂种类较多,具有不同的原理和特点,分析敏感性、特异性等性能指标也各有差异。国家和政府也应当建立相应的与国际标物溯源的标准物质、样本盘或者质控品,对已有的和不断产生的新的试剂进行分析敏感性、临床敏感性、重复性等性能指标进行评价。开展实验室间的室间质量评价,对实验室hpv检测或者基因分型的结果进行比对,以提高实验室的检测水平。本专利质控品的优势在于质控品简便易行,生产成本较低,非常适用于目前我国临床对hpvdna的检测评价。(1)准确性研究用hpv检测试剂盒检测本发明的质控品,得到的结果如表9,即3种hpv检测试剂盒检测结果表:表9(2)稳定性研究稳定性是体外诊断试剂必须具有的基本属性,是确保检测试剂在检测、使用过程中安全有效的重要指标。根据人乳头瘤病毒质控品的特性,设计合理的稳定性研究试验项目,以考察不同条件下质控品的主要质量指标随时间的变化情况,为质控品的保存条件和有效期的确定提供依据。a、运输稳定性:连续试生产3批质控品,按常规低温运输条件进行运输稳定性和运输后实时稳定性研究,每批均运输3天、4天、5天6天、7天、8天后,置于-15℃以下保存,然后在第3个月、第6个月、9个月、12个月、13个月、14个月进行质量检定。结果表明:本质控品经低温运输7天后,本质控品置于-15℃以下保存至14个月,经过质量检定完全符合质控品质量要求。根据质控品稳定性随时间递减的原则,确定质控品在途运输时间不超过7天;质控品的有效期为12个月,有效期内质控品性能稳定。先用水代替本质控品各组分,-15℃以下结冻,放进泡沫箱。在泡沫箱内放置冰袋后再装干冰,放温度记录仪进行温度监测,最后密封打包泡沫箱,并放进纸箱封箱。将泡沫箱进行运输监测,3kg干冰的的箱子,连续监测4天的温度变化;4kg干冰的箱子,则连续监测8天的温度变化。b、运输稳定性和实时稳定性试验根据模拟运输试验的结果,将质控品按照上述方法包装入泡沫箱,设置实验组,其中,a组为:装3kg干冰运输3天取出,置于-15℃以下保存;b组为:装3kg干冰运输4天取出,置于-15℃以下保存;c组为:装4kg干冰运输6天取出,置于-15℃以下保存;d组为:装4kg干冰运输7天取出,置于-15℃以下保存;e组为:装4kg干冰运输8天取出,置于-15℃以下保存;质检合格后分别在第3个月、第6个月、9个月、12个月、13个月、14个月对以上分组保存的质控品进行质量检定,以研究实时稳定性,得到的结果如表10所示。表10c、加速破坏稳定性:加速破坏性试验主要考察温度对质控品性能的破坏作用。取合格的质控品置于37℃恒温保存3天、5天、7天、9天,结果表明:本质控品在37℃保存5天性能稳定,至7天时性能开始下降。说明本质控品对温度的耐受性能力有限,须按照要求将本品置于-15℃以下保存。运输过程要求低温,同时使用干冰和冰袋。d、反复冻融稳定性:对已经过常规运输7天并检验合格的3批质控品进行研究,结果表明:本质控品中的反复冻融24次,经质量检定完全符合质控品质检要求。根据质控品稳定性随时间递减的原则,为保证质控品性能,确定本质控品反复冻融不应超过20次。稳定性研究结果件下表11。表11梯度1梯度2梯度3梯度4梯度5梯度6运输稳定性(天)345678-15℃保存(个月)691012141537℃恒温保存(天)013579反复冻融(次)151820222426实施例参见图1-8,图1-2为样本dna扩增后的pcr产物进行电泳的电泳结果图;图3为tvector的结构示意图(含克隆位点);图4为样本选择荧光图谱,图4中曲线由左至右依次对应标准品1、样品、标准品2、标准品3和标准品4;图5为标准曲线公式图;图6为稳定性研究起始ct值图;图7为稳定性研究ct值变化图;图8为数字pcr绝对定量法对23种质控品的定值结果图。本实施例的人乳头瘤病毒全长基因组质控品的制备方法包括:(1)hpv核酸提取及型别确定应用亚能生物技术(深圳)有限公司的核酸提取试剂,型号:病原体dna(离心柱型)来提取宫颈脱落细胞,具体方法是:充分洗脱宫颈刷,取0.5ml含样本的细胞保存液转移到1.5ml离心管中,13000rpm离心10分钟,吸弃300μl上清液(此时管内剩余液体和沉淀量大约为200μl);向细胞沉淀中加入20μlpde蛋白酶k,充分重悬细胞沉淀,低速离心数秒;再加入200μlpde裂解液,上下颠倒混匀,低速离心数秒;(注:不能将pde蛋白酶k直接加入到pde裂解液内)将盛放上述样本处理液的离心管置于56℃恒温裂解10min;裂解完成后,加入300μl无水乙醇,混匀,稍事离心。取pde离心柱置于pde收集管内,标记编号后,备用;转移1.5ml离心管内的样本处理液至标记好的pde离心柱内,盖上管盖,12000rpm离心1min。(注:转移样本处理液和加入洗液的过程中,应把液体对准pde离心柱中央缓缓加入,切勿将液体粘附在pde离心柱管口,以免影响dna的纯化质量);弃pde收集管内的滤液,将pde离心柱置回到pde收集管中,加入700μlpde洗液,盖上管盖,12000rpm离心1min;(注:为避免pde离心柱管口沾附的液体影响后续的洗液,可将弃滤液的pde收集管在纸巾上倒扣轻拍几下)弃pde收集管内的滤液,将pde离心柱置回到pde收集管中,加入700μl无水乙醇,盖上管盖,12000rpm离心1min;弃pde收集管内的滤液,将pde离心柱置回到pde收集管中,14000rpm空离心2min;弃pde收集管,将pde离心柱置于洁净的1.5ml离心管内,打开pde离心柱管盖,室温晾置5min;在pde离心柱的膜中央加入50μlpde洗脱液,盖上管盖,室温静置3min,8000rpm离心2min;弃pde离心柱,将提取的dna模板置于4℃(24h之内)或-18℃以下保存备用。应用亚能生物技术(深圳)有限公司的人乳头瘤病毒基因分型(23型)检测试剂盒(pcr-反向点杂交法)和人乳头瘤病毒核酸检测试剂盒(pcr-荧光探针法)检测提取的dna模版,得到的结果如表12所示,表12为hpv核酸提取及型别确定表。表12(2)获得目的片段应用上述pcr反应体系,扩增样本dna,使用1xtbe缓冲液制作浓度为1%的琼脂糖凝胶,然后对pcr产物进行电泳,电泳程序为170v5min,转100v80min,得到的电泳结果如图1-2所示。(3)回收目的片段应用琼脂糖凝胶回收纯化dna试剂盒来回收目的dna,具体操作为:在紫外灯下切出含有目的dna的琼脂糖凝胶,约0.2mg,用纸巾吸弃表面液体,按照4个凝胶体积量加入buffergm,混匀后先置于37℃加热10min,然后室温(15-25℃)溶解胶块,其间间断振荡混匀,使胶块充分溶解。将spincolumn安置于collectiontube上,将溶解液转移至tube的中央,注意一定要垂直滴加,12000rpm离心1min,并将滤液再次加入column中离心一次,提高dna回收效率。将700μl的bufferwb加入到column中,室温12000rpm离心30s,并重复一次,然后将column置于新的离心管上,室温开盖放置2min,再在中央处加入30μl已预热至60℃的tebuffer,室温静置1min,然后室温12000rpm离心1min洗脱dna。(4)载体与目的片段连接利用连接功能酶的特点,将dna片段和载体连接,构成hpv全基因组质粒dna。通常情况下,无论片段大小,载体和插入片段分别添加50-200ng就可达到较高的克隆效率。插入片段和载体的摩尔比例为2:1,如果pcr产物连接效率不够,可优化连接比例3:1至8:1。将反应体系置于50℃孵育15min,然后置于冰上放置5min即可连接成功。从-70℃冰箱中取dh5α感受态细胞100μl,放置于冰浴中融化,向感受态细胞悬液中加入已连接成功的质粒dna,轻弹混匀,在冰浴中静置30min,然后将离心管置于42℃水浴中放置60-90sec,然后快速将管转移到冰浴中,使细胞冷却2-3min,注意此过程中不能摇动离心管,然后向离心管中加入900μl无菌无抗生素的lb培养基,混匀后于37℃摇床振荡培养45min(150rpm),使质粒上相关的抗性标记基因表达,菌体复苏。然后再取100μl已转化的感受态细胞涂布在一个包含氨苄抗生素的lb平板上,氨苄抗生素浓度为100μg/ml,用无菌的弯头玻璃棒轻轻的将细胞均匀涂开。将平板置于室温直至液体被吸收,然后倒置于37℃过夜培养,次日挑取单克隆,扩大培养。(5)提取质粒,通过酶切处理、pcr筛选或者测序验证序列是否匹配,得到的结果如表4中对比序列id。应用快速质粒小提试剂盒(离心柱型)提取质粒,将4ml过夜培养的菌液加入离心管中,12000rpm离心1min,吸弃上清,加入150μl溶液p1,振荡混匀,测地悬浮细胞沉淀,此时溶液为浑浊的红色。然后加入150μl溶液p2,温和的上下颠倒混匀6-8次,注意不能剧烈振荡,以免污染基因组dna,此时菌液应变为清亮粘稠的紫色。向离心管中加入350μl溶液p5,立即快速地上下颠倒混匀12-20次,避免产生局部沉淀,充分混匀,此时将出现絮状沉淀,溶液为澄清的黄色,如果在黄色中混有紫色,则说明复性不充分,继续混匀至溶液颜色完全变为澄清的黄色。然后12000rpm离心2min,将上清液转移到吸附柱cp3中(吸附柱放入收集管中),注意尽量不要吸出沉淀。12000rpm离心30sec,倒掉收集管中的废液,将吸附柱cp3放入收集管中向吸附柱cp3中加入300μl漂洗液pwt(请先检查是否已加入无水乙醇),12000rpm离心30sec,倒掉收集管中的废液,将吸附柱cp3放入收集管中,将吸附柱cp3放入收集管中,12000rpm离心1min,去除残留的漂洗液。将吸附柱cp3置于一个干净的离心管中,向吸附膜的中间部位滴加50-100μl洗脱缓冲液tb,12000rpm离心30sec将质粒溶液收集到离心管中。(6)基本定值方法使用吸光度值法测量质粒原液的核酸浓度及纯度,然后按拷贝数计算公式算出23种hpv基因型质控品浓度,然后根据已有的高危型稀释倍数及拷贝数计算公式推出23种hpv基因型质控品浓度为105-107copies/ml。汇总结果见表13。表13为用吸光度值法对23种质控品的定值结果表。表13用人乳头瘤病毒核酸检测试剂盒(pcr-荧光探针法)对中国食品药品检定研究院研制体外诊断试剂国家标准物质人乳头瘤病毒全基因组分型参考品的5个浓度进行3次重复检测(结果分析时取3次结果的平均值),建立梯度稀释结果的标准曲线,再将人乳头瘤病毒核酸检测试剂盒(pcr-荧光探针法)检测12种hpv型(分别为:hpv16、18、26、31、33、35、45、56、58、59、66、82)的检测结果(结果分析时取3次结果的平均值)代入各基因型的标准曲线方程中,得出浓度范围为105-107copies/ml。12种国参的浓度梯度检测结果如表14所示,12种质控品与国参溯源的定值结果如表15所示。表14表15通过与中国食品药品检定研究院研制体外诊断试剂国家标准物质人乳头瘤病毒全基因组分型参考品对标以及吸光度值法与拷贝数的推断方法,对23种hpv阳性质控品进行定值研究,所有质控品的浓度范围为:105-107copies/ml。(7)绝对定量方法使用数字pcr绝对定量法,简易操作流程为:1)按表6的配方配制24人份数字pcr反应液并加入质控品1至质控品23;2)正确放置数字pcr芯片、刷头及芯片盖;3)将已加完样的反应液全部加入刷头下部的加样孔,注意不要有气泡,然后刷头会将反应液均匀的涂抹在芯片上。待涂抹完成后,加入10-15滴密封油,使密封油能够覆盖芯片表面,盖上芯片盖,紧密的按压至少15秒后取出芯片,按表7的扩增程序扩增,大约2小时30分钟后,取出扩增完成的芯片,并放入读取仪中,读取仪会自动阅读质控品的目的dna浓度。用数字pcr绝对定量法对23种质控品的定值结果的汇总结果见表16以及图8。表16通过数字pcr的方法对23种hpv阳性质控品进行定值研究,所有质控品在稀释10倍后,加入量为2μl时,绝对定量的结果为100至400拷贝,即5x105-2x106copies/ml的浓度范围。基于上述,本发明是将23个型的人乳头瘤病毒全基因组dna克隆成质粒dna,从而实现对hpv基因检测试剂盒的质量控制和评价;包括:(1)利用高保真dna聚合酶的特性,扩增出hpv全基因组dna片段,建立了一种可将8000bp的片段整体高保真高效率扩增的方法;(2)利用hdcloning酶的连接特性,将目的片段无缝、定向的克隆至载体的目的位置,避免连接效率低下的问题;(3)利用数字pcr绝对定量技术,确定了质粒dna的浓度,解决了质控品浓度不确定的问题;(4)成功地得到无生物传染危险型而又可模拟临床样本、可大量生产、浓度可控、稳定保存的hpv基因检测质控品,具有较好的适用性。综上所述,本发明提供的人乳头瘤病毒全长基因组质控品的制备方法可成功地得到无生物传染危险型而又可模拟临床样本、可大量生产、浓度可控、稳定保存的hpv基因检测质控品,具有较好的适用性。以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的
技术领域:
,均同理包括在本发明的专利保护范围内。sequencelisting<110>亚能生物技术(深圳)有限公司<120>hpv全长基因组质控品的制备方法、扩增引物及检测试剂<130>2018<160>51<170>patentinversion3.5<210>1<211>40<212>dna<213>人工序列<400>1acccggggatccgattgttaataacaatcttggtttaaaa40<210>2<211>40<212>dna<213>人工序列<400>2acccggggatccgattcttaataacaatcttagtttaaaa40<210>3<211>40<212>dna<213>人工序列<400>3acccggggatccgattggagtgaccgaaaacggtcgtacc40<210>4<211>40<212>dna<213>人工序列<400>4acccggggatccgattttgtttaaaaaaagggagtaaccg40<210>5<211>40<212>dna<213>人工序列<400>5acccggggatccgatttaacaattatatgttactaaaagg40<210>6<211>38<212>dna<213>人工序列<400>6acccggggatccgattgttaagaccgaaaacggtgcat38<210>7<211>40<212>dna<213>人工序列<400>7acccggggatccgatttgtaaccgaaaacggtttcaaccg40<210>8<211>40<212>dna<213>人工序列<400>8acccggggatccgatttgtaaccgaaaacggtttcaaccg40<210>9<211>40<212>dna<213>人工序列<400>9acccggggatccgatttataataaagtagggaggtaccga40<210>10<211>40<212>dna<213>人工序列<400>10acccggggatccgattacaattatcttgtaaaaagtaggg40<210>11<211>40<212>dna<213>人工序列<400>11acccggggatccgatttaacaattatatgttactaaaagg40<210>12<211>40<212>dna<213>人工序列<400>12acccggggatccgattaaaaagtagggagtgaccgaaagt40<210>13<211>40<212>dna<213>人工序列<400>13acccggggatccgattggagtgaccgaaaacggtcgtacc40<210>14<211>40<212>dna<213>人工序列<400>14acccggggatccgattttgtttaaaaaaagggagtaaccg40<210>15<211>40<212>dna<213>人工序列<400>15acccggggatccgattctaaactataatgccaaatcttgt40<210>16<211>38<212>dna<213>人工序列<400>16acccggggatccgattgttaagaccgaaaacggtgcat38<210>17<211>40<212>dna<213>人工序列<400>17acccggggatccgatttgtaaccgaaaacggtttcaaccg40<210>18<211>40<212>dna<213>人工序列<400>18acccggggatccgatttgtaaccgaaaacggtttcaaccg40<210>19<211>40<212>dna<213>人工序列<400>19acccggggatccgatttataataaagtagggaggtaccga40<210>20<211>40<212>dna<213>人工序列<400>20acccggggatccgattacaattatcttgtaaaaagtaggg40<210>21<211>40<212>dna<213>人工序列<400>21acccggggatccgatttaacaattatatgttactaaaagg40<210>22<211>40<212>dna<213>人工序列<400>22acccggggatccgattaaaaagtagggagtgaccgaaagt40<210>23<211>40<212>dna<213>人工序列<400>23acccggggatccgattggagtgaccgaaaacggtcgtacc40<210>24<211>40<212>dna<213>人工序列<400>24actagtcatatggattataagaaggaaatatgtagggtgt40<210>25<211>40<212>dna<213>人工序列<400>25actagtcatatggatttacaagtacacacgcatacataaa40<210>26<211>40<212>dna<213>人工序列<400>26actagtcatatggattacattgcacaatattatacacaca40<210>27<211>40<212>dna<213>人工序列<400>27actagtcatatggattgaaatgttgcagacctattatatg40<210>28<211>40<212>dna<213>人工序列<400>28actagtcatatggattaacacgcctttacattacatacag40<210>29<211>40<212>dna<213>人工序列<400>29actagtcatatggattaaaacaaatacacacatacagcag40<210>30<211>40<212>dna<213>人工序列<400>30actagtcatatggattgtcaagtaaaccacccacacaaca40<210>31<211>40<212>dna<213>人工序列<400>31actagtcatatggattacacccaggtttggaacagagcat40<210>32<211>40<212>dna<213>人工序列<400>32actagtcatatggatttaaaccacacagaaaccatacagc40<210>33<211>40<212>dna<213>人工序列<400>33actagtcatatggattaacacgcctttacattacatacag40<210>34<211>40<212>dna<213>人工序列<400>34actagtcatatggatttacaagtacacacgcatacataaa40<210>35<211>40<212>dna<213>人工序列<400>35actagtcatatggattacattgcacaatattatacacaca40<210>36<211>40<212>dna<213>人工序列<400>36actagtcatatggattgaaatgttgcagacctattatatg40<210>37<211>40<212>dna<213>人工序列<400>37actagtcatatggattggaaactgacaaggacatagaaca40<210>38<211>40<212>dna<213>人工序列<400>38actagtcatatggattaaaacaaatacacacatacagcag40<210>39<211>40<212>dna<213>人工序列<400>39actagtcatatggattgtcaagtaaaccacccacacaaca40<210>40<211>40<212>dna<213>人工序列<400>40actagtcatatggattacacccaggtttggaacagagcat40<210>41<211>40<212>dna<213>人工序列<400>41actagtcatatggatttaaaccacacagaaaccatacagc40<210>42<211>40<212>dna<213>人工序列<400>42actagtcatatggattaacacgcctttacattacatacag40<210>43<211>40<212>dna<213>人工序列<400>43actagtcatatggatttacaagtacacacgcatacataaa40<210>44<211>40<212>dna<213>人工序列<400>44actagtcatatggattacattgcacaatattatacacaca40<210>45<211>40<212>dna<213>人工序列<400>45actagtcatatggattgaaatgttgcagacctattatatg40<210>46<211>40<212>dna<213>人工序列<400>46actagtcatatggattggaaactgacaaggacatagaaca40<210>47<211>25<212>dna<213>人工序列<400>47gaaaaataaactgtaaatgatattc25<210>48<211>25<212>dna<213>人工序列<400>48gaaaaataaactgtaaatcatattc25<210>49<211>20<212>dna<213>人工序列<400>49gcmcagggwcataayaatgg20<210>50<211>20<212>dna<213>人工序列<400>50gcmcagggwcacaayaatgg20<210>51<211>22<212>dna<213>人工序列<400>51catttgctggggtaaccaacta22当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1