红色磷光化合物和使用该化合物的有机电致发光器件的制作方法
本发明涉及一种有机电致发光器件,具体而言,涉及红色磷光化合物和使用它的有机电致发光器件。特别地,本发明涉及用作有机电致发光器件的发光层的掺杂剂的红色磷光体。
背景技术:
近年来,随着显示器件的尺寸越来越大,越来越需要占用较少空间的平板显示器件。该平板显示器件包括有机电致发光器件,也称为有机发光二极管(oled)。该有机电致发光器件的技术正在以巨大的速度发展,已经公开了许多原型。
当电荷注入到电子注入电极(阴极)和空穴注入电极(阳极)之间形成的有机层中时,有机电致发光器件发光。更具体地,当电子和空穴形成一对时发光,新产生的电子空穴对衰减。可以在柔性透明基板如塑料上形成有机电致发光器件。也可以在比等离子体显示板或无机电致发光(el)显示器中需要的电压更低的电压(即低于或等于10v的电压)下驱动有机电致发光器件。有机电致发光器件的有利之处在于,其与其他显示器件相比消耗更少的能量和提供优异的颜色显示。而且,由于有机电致发光器件使用三色(即绿色,蓝色和红色),可以再现图片,所以有机电致发光器件被广泛地认为是可以再现清晰图像的下一代颜色显示器件。
制造有机发光二极管(el)器件的过程描述如下:
(1)将阳极材料涂布在透明基板上。一般使用氧化铟锡(ito)作阳极材料。
(2)在阳极材料上沉积空穴注入层(hil)。空穴注入层由具有10纳米(nm)至30(nm)厚度的铜酞菁(cupc)层形成。
(3)然后沉积空虚传输层(htl)。空穴传输层主要由4,4’-双[n-(1-萘基)-n-苯氨基]联苯(npb)形成,先用真空蒸发处理,然后涂布至具有30纳米(nm)至60纳米(nm)的厚度。
(4)此后,形成有机发光层。此时,如果需要,可以加入掺杂剂。在发绿光的情况中,有机发光层一般由真空蒸发至具有30纳米(nm)至60纳米(nm)厚度的三(8-羟基喹啉酸)铝(alq3)形成。并且使用,mqd(n-甲基喹吖啶铜)作为掺杂剂(或杂质)。
(5)在有机发光层上依次形成电子传输层(etl)和电子注入层(eil),或在有机发光层上形成电子注入/传输层。在发绿光的情况中,步骤(4)的alq3具有优异的电子传输能力。因此,并非必须地需要电子注入和传输层。
(6)最后涂布阴极层,在整个结构上涂布保护层。
依照在上述结构中形成发光层的方法,决定了分别发出(或显示)蓝,绿,红颜色的发光器件。作为发光材料,由从每个电极注入的电子和空穴的重组而形成激子。单线态激子发射荧光,三线态激子发射磷光。发射荧光的单线态激子具有25%的形成可能性,而发射磷光的三线态激子具有75%的形成可能性。因此,与单线态激子相比,三线态激子提供更大的发光效率。在这样的磷光材料中,红色磷光材料比荧光材料可以具有更大的发光效率。因此,作为提高有机电致发光器件的效率的重要因素,红色磷光材料正在被广泛研究。
当使用这样的磷光材料时,需要高发光效率,高色纯度和延长的耐久性。最特别地,当使用红色磷光材料时,随色纯度增加(即cie色度坐标的x值变得更大),可见度降低,从而导致难以提供高发光效率。因此,需要开发能提供优异的色度坐标(x的cie色纯度=0.63以上),提高的发光效率和延长的耐久性的红色磷光材料。
技术实现要素:
本发明致力于提供红色磷光化合物和使用该红色磷光化合物的有机电致发光器件,其基本消除了由于相关技术的限制和缺点而导致的一个或多个问题。
本发明的一个目的在于,提供一种化合物作为有机电致发光器件的发光层中作为掺杂剂,从而提供具有高色纯度、高亮度和长耐久性的有机电致发光器,其结构式如i所示,
其中,
其中,r1、r2、r3和r4独立选自h、c1~c6烷基的一种。
且其中,
优选的,所述c1~c6烷基选自甲基,甲基-d3,乙基,正丙基,异丙基,正丁基,异丁基和叔丁基中的一种。
具体而言,式i可以是下列化学式中的任一个:
本发明的另一个目的在于提供一种有机发光二极管器件,包括彼此顺次沉积的阳极,空穴注入层,空穴传输层,发光层,电子传输层,电子注入层和阴极的有机电致发光器件,所述有机发光二极管器件包含上述任何一种红色磷光化合物作为掺杂剂。本发明提供的红色磷光化合物可以加在任一层做掺杂剂,但主要把该化合物运用在发光层做掺杂剂。
优选的,所述有机发光二极管器件中使用al和zn金属络合物和咔唑衍生物中的任一种作为发光层的主体材料,掺杂剂的用量范围可以在0.1重量%~50重量%范围内。当使用的掺杂剂的量在上述范围内时,可以提高本发明的效率。
优选的,所述al或zn金属络合物的配体为喹啉基、联苯基、异喹啉基、苯基、甲基喹啉基、二甲基喹啉基、二甲基异喹啉基中的一种或几种;所述咔唑衍生物为4,4’-n,n’-二咔哇联苯(cbp)。
本发明提供的红色磷光化合物有益效果在于能使有机电致发光器件具有高效率和高色纯度和窄光谱。
附图说明
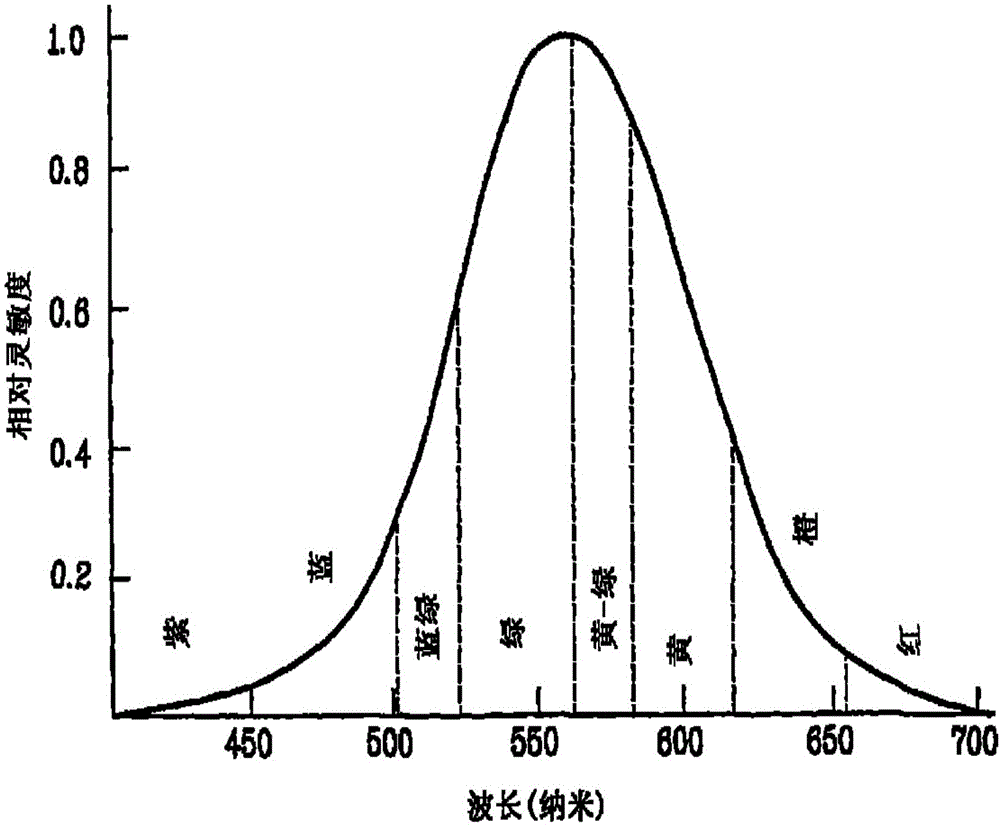
图1为波长与相对灵敏度的关系的图。
具体实施方式
下文给出优选实施方案的例子以描述本发明。显然应理解的是,本发明不仅仅局限于所提出的实施方案。
由于结构式如i的红色磷光化合物均为提供优异的色度坐标(x的cie色纯度=0.64以上),提高的发光效率和延长的耐久性的红色磷光材料,现以rd-001、rd-361、rd-002、rd-362、rd-006和rd-366制备方法和测试结果为例,证明本发明提供的技术方案和达到的技术效果。
图1说明表示随有机电致发光器件的色纯度增加(即随色度坐标的x值变得更大)可见度降低的图。
以下实施方案中,npb为4,4’-双[n-(1-萘基)-n-苯氨基]联苯,cbp为4,4’-n,n’-二咔哇联苯,cupc为酞菁铜,lif氟化锂,ito为氧化铟锡,alq3为三(8-羟基喹啉)铝。
lc-ms:液相色谱-质谱联用仪,m/z:质子数/电荷数的比值。
下图为在本发明的实施方案中使用的化合物铜(ⅱ)酞菁(cupc),npb,(btp)2ir(acac),alq3和cbp的结构式。
1.7-异丙基异喹啉的合成
在氮气保护条件下,向三口瓶中加入无水氯化锌78.6克(0.58mol),四氢呋喃800ml,降低反应体系温度至0℃,滴加异丙基氯化镁480ml(0.96mol),保温搅拌一个半小时。之后加入双苯基膦二茂铁二氯化钯3.5克(1mol%),搅拌15分钟,滴加7-溴异喹啉100克(0.48mol).反应12小时后停止。用乙酸乙酯和水萃取反应液,干燥浓缩。浓缩得到的晶体经过重结晶纯化获得61.6克(收率:75%)7-异丙基异喹啉。lc-ms:m/z172.2(m+h)+
2.7-溴异喹啉2-氧化物的合成
在三颈瓶中将7-异丙基异喹啉61克(0.36mol),溶于醋酸400ml,缓慢加入双氧水200ml。缓慢升温反应体系至80℃搅拌回流。回流反应12h后停止反应。蒸馏除去醋酸,用乙酸乙酯与水萃取反应混合物,取有机相用无水硫酸镁干燥。干燥后的有机相蒸干,粗产物通过四氢呋喃与甲醇溶剂重结晶分离提纯得到最终产物47.9克(收率:60%)7-溴异喹啉2-氧化物。
lc-ms:m/z225.1(m+h)+
3.1-氯-7-异丙基异喹啉的合成
在三颈瓶中加入7-溴异喹啉2-氧化物47克(0.21mol),三氯氧磷41.8克(0.27mol)和二氯甲烷400ml。升温反应体系至50℃回流。回流反应6h后停止反应。蒸馏除去醋酸,用乙酸乙酯与水萃取反应混合物,取有机相用无水硫酸镁干燥。将干燥后的有机相浓缩,粗产物以乙酸乙酯和正己烷1:40的混合溶剂通过层析硅胶柱分离提纯得到产物30.2克(收率:70%)1-氯-7-异丙基异喹。lc-ms:m/z206.7(m+h)+
4.环己烯-1-硼酸频哪醇酯的合成
在氮气保护条件下,向三口瓶中加入1-溴-1-环己烯20克(0.12mol),联硼酸频那醇酯34.7克(0.14mol),三苯基膦2.0克(6mol%),反-二(三苯基膦)二氯化钯(ii)2.6克(3mol%),苯酚钾24.6克(0.19mol)和无水甲苯250ml。氮气置换后在50℃条件下搅拌反应5小时,然后将体系冷却到室温并加水猝灭反应。反应混合物用苯溶剂和饱和食盐水萃取。取有机相用无水硫酸镁干燥。将干燥后混合物过滤并减压浓缩,可通过硅胶层析柱或者蒸馏法进行提纯得到21.9克(收率:85%)环己烯-1-硼酸频哪醇酯。lc-ms:m/z209.1(m+h)+
5.3-甲基环己烯-1-硼酸频哪醇酯的合成
在氮气保护条件下,向三口瓶中加入1-溴-3-甲基-1-环己烯15克(85.7mmol),联硼酸频那醇酯23.9克(94.2mmol),三苯基膦1.4克(6mol%),反-二(三苯基膦)二氯化钯(ii)1.8克(3mol%),苯酚钾20.0克(128.5mol)和无水甲苯200ml。氮气置换后在50℃条件下搅拌反应5小时,然后将体系冷却到室温并加水猝灭反应。反应混合物用苯溶剂和饱和食盐水萃取。取有机相用无水硫酸镁干燥。将干燥后混合物过滤并减压浓缩,可通过硅胶层析柱或者蒸馏法进行提纯得到15.6克(收率:82%)3-甲基环己烯-1-硼酸频哪醇酯。
lc-ms:m/z223.1(m+h)+
6.3,5-二甲基环己烯-1-硼酸频哪醇酯的合成
在氮气保护条件下,向三口瓶中加入1-溴-3,5-二甲基-1-环己烯15克(79.3mmol),联硼酸频那醇酯34.7克(87.26mmol),三苯基膦1.3克(6mol%),反-二(三苯基膦)二氯化钯(ii)1.7克(3mol%),苯酚钾15.7克(119mmol)和无水甲苯200ml。氮气置换后在50℃条件下搅拌反应5小时,然后将体系冷却到室温并加水猝灭反应。反应混合物用苯溶剂和饱和食盐水萃取。取有机相用无水硫酸镁干燥。将干燥后混合物过滤并减压浓缩,可通过硅胶层析柱或者蒸馏法进行提纯得到14.1克(收率:75%)3,5-二甲基环己烯-1-硼酸频哪醇酯。lc-ms:m/z237.2(m+h)+
7.1-(环己-1-烯-1-基)-7-异丙基异喹啉的合成
在氮气保护下,向三口瓶中加入1-氯-6-异丙基异喹啉10克(48.6mmol),环己烯-1-硼酸频哪醇酯11.1克(53.5mmol),2m-碳酸钾80ml溶于四氢呋喃80ml中.氮气置换30分钟,加入催化剂四三苯基膦钯(1mol%)。将反应体系升温至80℃,搅拌回流12小时。冷却至室温,加水猝灭反应,用乙酸乙酯和饱和食盐水萃取反应液。用饱和食盐水洗涤两到三次,取有机相。有机相用无水硫酸镁干燥后浓缩。通过硅胶层析柱分离提纯可得到9.8克(收率:80%)1-(环己-1-烯-1-基)-7-异丙基异喹啉。lc-ms:m/z252.4(m+h)+
8.7-异丙基-1-(3-甲基环己-1-烯-1-基)异喹啉的合成
在氮气保护下,向三口瓶中加入1-氯-6-异丙基异喹啉10克(48.6mmol),3-甲基环己烯-1-硼酸频哪醇酯11.9克(53.5mmol),2m-碳酸钾80ml溶于四氢呋喃80ml中.氮气置换30分钟,加入催化剂四三苯基膦钯(1mol%)。将反应体系升温至80℃,搅拌回流12小时。冷却至室温,加水猝灭反应,用乙酸乙酯和饱和食盐水萃取反应液。用饱和食盐水洗涤两到三次,取有机相。有机相用无水硫酸镁干燥后浓缩。通过硅胶层析柱分离提纯可得到9.8克(收率:76%)7-异丙基-1-(3-甲基环己-1-烯-1-基)异喹啉。lc-ms:m/z266.4(m+h)+
9.1-(3,5-二甲基丙-1-烯-1-基)-7-异丙基异喹啉的合成
在氮气保护下,向三口瓶中加入1-氯-6-异丙基异喹啉10克(48.6mmol),3,5-二甲基环己烯-1-硼酸频哪醇酯12.6克(53.5mmol),2m-碳酸钾60ml溶于四氢呋喃60ml中.氮气置换30分钟,加入催化剂四三苯基膦钯(1mol%)。将反应体系升温至80℃,搅拌回流12小时。冷却至室温,加水猝灭反应,用乙酸乙酯和饱和食盐水萃取反应液。用饱和食盐水洗涤两到三次,取有机相。有机相用无水硫酸镁干燥后浓缩。通过硅胶层析柱分离提纯可得到10.2克(收率:75%)1-(3,5-二甲基丙-1-烯-1-基)-7-异丙基异喹啉。
lc-ms:m/z280.4(m+h)+
10.二氯交联二聚体配合物的合成
将三氯化铱的一水合物4克(13.4mmol),1-(环己-1-烯-1-基)-7-异丙基异喹啉7.4克(29.5mmol)和二乙醇单乙醚与蒸馏水的比例为3/1(120ml/40ml)的混合溶液,加入干燥的两口圆底烧瓶中,加热回流反应24小时,然后加入适量的蒸馏水,再将析出的固体过滤,并用甲醇和石油醚洗涤固体,得到6.2克(收率:64%)二氯交联二聚体配合物。lc-ms:m/z1457.8(m+h)+
11.rd-001的合成
将二氯交联二聚体配合物5克(3.4mmol),戊烷-2,4-二酮1.7克(17.2mmol),无水碳酸钠2.2克(20.6mmol)和2-乙氧基乙醇80ml加入双口圆底烧瓶中,然后加热回流反应6小时,停止加热,降至室温,加入适量的蒸馏水,过滤出固体。将固体溶解在二氯甲烷中,过硅胶短柱。在减压条件下除去溶剂,浓缩得到的固体先后用甲醇和石油醚洗涤,得到4.1克(收率:75%)所需产物。lc-ms:m/z793.0(m+h)+
12.rd-361的合成
将二氯交联二聚体配合物5克(3.4mmol),3,7-二乙基-5-甲基-4,6-壬二酮2.2克(10.3mmol),无水碳酸钠2.2克(20.1mmol)和2-乙氧基乙醇80ml加入双口圆底烧瓶中,然后加热回流反应6小时,停止加热,降至室温,加入适量的蒸馏水,过滤出固体。将固体溶解在二氯甲烷中,过硅胶短柱。在减压条件下除去溶剂,浓缩得到的固体先后用甲醇和石油醚洗涤,得到4.5克(收率:72%)所需产物。lc-ms:m/z905.3(m+h)+
13.二氯交联二聚体配合物的合成
将三氯化铱的一水合物4克(13.4mmol),7-异丙基-1-(3-甲基环己-1-烯-1-基)异喹啉7.8克(29.5mmol)和二乙醇单乙醚与蒸馏水的比例为3/1(120ml/40ml)的混合溶液,加入干燥的两口圆底烧瓶中,加热回流反应24小时,然后加入适量的蒸馏水,再将析出的固体过滤,并用甲醇和石油醚洗涤固体,得到6.1克(收率:60%)二氯交联二聚体配合物。lc-ms:m/z1513.9.0(m+h)+
14.rd-002的合成
将二氯交联二聚体配合物5克(3.3mmol),戊烷-2,4-二酮1.7克(16.5mmol),无水碳酸钠2.1克(19.8mmol)和2-乙氧基乙醇80ml加入双口圆底烧瓶中,然后加热回流反应6小时,停止加热,降至室温,加入适量的蒸馏水,过滤出固体。将固体溶解在二氯甲烷中,过硅胶短柱。在减压条件下除去溶剂,浓缩得到的固体先后用甲醇和石油醚洗涤,得到3.8克(收率:70%)所需产物。lc-ms:m/z821.1(m+h)+
15.rd-362的合成
将二氯交联二聚体配合物5克(3.3mmol),3,7-二乙基-5-甲基-4,6-壬二酮2.1克(9.9mmol),无水碳酸钠2.1克(19.8mmol)和2-乙氧基乙醇80ml加入双口圆底烧瓶中,然后加热回流反应6小时,停止加热,降至室温,加入适量的蒸馏水,过滤出固体。将固体溶解在二氯甲烷中,过硅胶短柱。在减压条件下除去溶剂,浓缩得到的固体先后用甲醇和石油醚洗涤,得到4.0克(收率:65%)所需产物。lc-ms:m/z933.3(m+h)+
16.二氯交联二聚体配合物的合成的合成
将三氯化铱的一水合物4克(13.4mmol),1-(3,5-二甲基丙-1-烯-1-基)-7-异丙基异喹啉8.2克(29.5mmol)和二乙醇单乙醚与蒸馏水的比例为3/1(120ml/40ml)的混合溶液,加入干燥的两口圆底烧瓶中,加热回流反应24小时,然后加入适量的蒸馏水,再将析出的固体过滤,并用甲醇和石油醚洗涤固体,得到6.3克(收率:60%)二氯交联二聚体配合物。
lc-ms:m/z1570.0(m+h)+
17.rd-006的合成
将二氯交联二聚体配合物5克(3.2mmol),戊烷-2,4-二酮1.6克(15.9mmol),无水碳酸钠2.0克(19.2mmol)和2-乙氧基乙醇80ml加入双口圆底烧瓶中,然后加热回流反应6小时,停止加热,降至室温,加入适量的蒸馏水,过滤出固体。将固体溶解在二氯甲烷中,过硅胶短柱。在减压条件下除去溶剂,浓缩得到的固体先后用甲醇和石油醚洗涤,得到3.5克(收率:65%)所需产物。lc-ms:m/z849.2(m+h)+
18.rd-366的合成
将二氯交联二聚体配合物5克(3.2mmol),3,7-二乙基-5-甲基-4,6-壬二酮2.0克(9.6mmol),无水碳酸钠2.0克(19.2mmol)和2-乙氧基乙醇80ml加入双口圆底烧瓶中,然后加热回流反应6小时,停止加热,降至室温,加入适量的蒸馏水,过滤出固体。将固体溶解在二氯甲烷中,过硅胶短柱。在减压条件下除去溶剂,浓缩得到的固体先后用甲醇和石油醚洗涤,得到3.7克(收率:60%)所需产物。lc-ms:m/z961.4(m+h)+
实施方案
1.第一实施方案
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。此后,在ito基板上以cupc
在0.9ma下,亮度等于1153cd/m2(6.1v)。此时,ciex=0.674,y=0.328。
2.第二实施方案
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。此后,在ito基板上以cupc
在0.9ma下,亮度等于1228cd/m2(6.3v)。此时,ciex=0.675,y=0.326。
3.第三实施方案
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。此后,在ito基板上以cupc
在0.9ma下,亮度等于1119cd/m2(6.2v)。此时,ciex=0.677,y=0.325。
4.第四实施方案
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。此后,在ito基板上以cupc
在0.9ma下,亮度等于1173cd/m2(6.4v)。此时,ciex=0.679,y=0.323。
5.第五实施方案
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。此后,在ito基板上以cupc
在0.9ma下,亮度等于1102cd/m2(6.1v)。此时,ciex=0.682,y=0.322。
6.第六实施方案
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。此后,在ito基板上以cupc
在0.9ma下,亮度等于1148cd/m2(6.4v)。此时,ciex=0.684,y=0.319。
7.对比例
使ito玻璃基板图案化,以具有3mm×3mm的发光区域。然后,洗涤图案化的ito玻璃基板。随后将该基板安放在真空室。标准压力设定为1×10-6托。在ito基板上以cupc
当使用balq形成空穴负载层时,在0.9ma下,亮度等于689cd/m2(8.1v)。此时,ciex=0.651,y=0.329。
依据上述实施方案和对比例,效率,色度坐标,和亮度的特性显示在下表1。
表1
如表1所示,甚至当色纯度高时,该器件在低电压下也高效率地运行。并且,与对比例相比,第二实施方案的电流效率增加100%以上。通过使用式ⅰ所示的化合物作为有机电致发光器件的发光层,本发明提供具有优异的色纯度和亮度以及延长的耐久性的有机电致发光器件。
本领域的技术人员将明显看到,在不脱离本发明的精神和范围下,本发明可以有许多修改和变化。因此可以预期到,本发明覆盖在附带权利要求的范围及其相当范围内提供的本发明的修改和变化。
- 还没有人留言评论。精彩留言会获得点赞!