一种阿哌沙班有关物质及其制备方法和用途与流程
本发明涉及医药制造
技术领域:
,具体涉及一种阿哌沙班有关物质及其制备方法和用途。
背景技术:
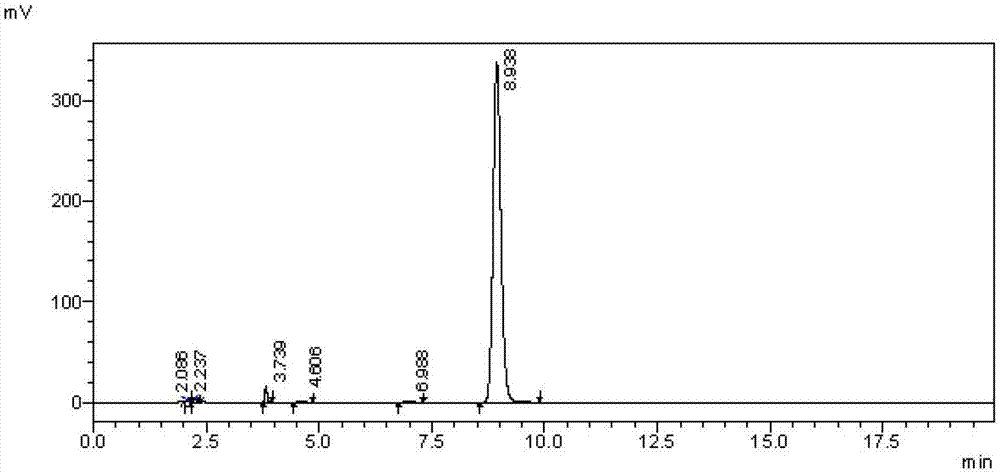
:阿哌沙班(apixaban),分子式为c25h25n5o4,其化学名为4,5,6,7-四氢-1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代-1-哌啶基)苯基]-1h-唑唑并[3,4-c]吡啶-3-羰胺。阿哌沙班是一种新型的xa因子抑制剂,由辉瑞与百时美施贵宝联合开发,用于预防成人全髋或全膝关节置换术后静脉血栓栓塞症,预防非瓣膜性房颤患者卒中和体循环栓塞,治疗及预防深静脉血栓栓塞症和肺栓塞。wo2017187245、us20150353543、wo2014075648和cn105254630等专利文献报道了阿哌沙班的合成,其中最后一步的合成方法如下:发明人在采用文献路线,如wo2017187245、us20150353543和wo2014075648等公开的方法进行阿哌沙班最后一步合成时,发现终产品中含有峰面积大于0.1%的杂质,并且该杂质在阿哌沙班成品中难以去除。因此,确认该杂质的化学结构以及制备方法,对建立检测方法,从而进行阿哌沙班的质量控制以及临床用药安全监测均起到了至关重要的作用。技术实现要素:本发明的目的在于克服上述技术不足,提供一种阿哌沙班有关物质,以便对阿哌沙班的合成过程进行质量控制;本发明第二方面的目的在于,提供一种阿哌沙班有关物质的制备方法,以便于制备高纯度的杂质对照品;本发明第三方面的目的在于,提供一种阿哌沙班有关物质的用途。为达到上述技术目的,本发明的技术方案提供一种阿哌沙班有关物质a,其具有如下结构:分子式:c25h26n4o6分子量:478.51。本发明的技术方案还提供了一种阿哌沙班有关物质a的制备方法,其合成路线如下:包括如下步骤:s1.在碱性条件下,化合物ii与5-氯戊酸乙酯发生亲核取代反应得到中间体iii;s2.所述中间体iii在强碱作用下,水解反应得到化合物a。本发明的技术方案还提供了一种阿哌沙班有关物质a的应用,其在阿哌沙班质量控制中作为杂质对照品。与现有技术相比,本发明的有益效果包括:1、本发明提供了一种阿哌沙班相关物质a,并对该物质的结构进行了确证,以克服现有阿哌沙班制备过程中杂质不易控制的缺点;2、本发明还提供了一种阿哌沙班相关物质a的制备方法,该方法制备过程反应条件温和、可控性好、产率较高、纯度高,适合工业化生产;3、本发明提供的阿哌沙班相关物质a可以作为标准品使用,以便对阿哌沙班制备过程中的质量进行控制研究。附图说明图1为阿哌沙班粗品的hplc图谱;图2为实施例4中制备的化合物a的hplc图谱。具体实施方式本实施例提供了一种阿哌沙班有关物质a的制备方法,包括如下步骤:(1)在第一有机溶剂中,碱性条件下,化合物ii(化学名为:1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代哌啶-1-基)苯基]-4,5,6,7-四氢-1h-吡唑并[3,4-c]吡啶-3-羧酸乙酯)与5-氯戊酸乙酯在0~100℃下发生亲核取代反应,反应至化合物ii消失,向反应液中加入适量水析出固体后,抽滤,滤饼用水和异丙醇洗涤,再经柱层析纯化后,得到中间体iii;(2)将中间体iii溶于第二有机溶剂中,在冰浴下向反应液中加入强碱溶液,将反应液的ph调至10~11,加完后,将反应液的温度调至0~40℃,水解反应直至中间体iii消失,将反应体系温度调至0℃,并将反应体系的ph调至4~5,析出固体后,再依次经过抽滤、干燥和重结晶,得到阿哌沙班有关物质a。在一些优选实施方式中,化合物ii、碱和5-氯戊酸乙酯的摩尔比为1:1~3:1~1.5,更优选的,化合物ii、碱和5-氯戊酸乙酯的摩尔比为1:1.2~2:1.1~1.2,在该用量比范围内,能保证亲核取代反应更充分的进行,并避免了原料的浪费。在一些优选实施方式中,每克化合物ii中加入5~30ml第一有机溶剂,更优选的,每克化合物ii中加入6~10ml第一有机溶剂,在该用量比范围内,能使化合物ii完全溶于第一有机溶剂中,使化合物ii、碱和5-氯戊酸乙酯充分接触。在一些优选实施方式中,第一有机溶剂为四氢呋喃、乙腈、n,n-二甲基甲酰胺和n-甲基吡咯烷酮中的一种或几种,更优选的,第一有机溶剂为n,n-二甲基甲酰胺。在一些优选实施方式中,步骤s1中向反应体系中加入的碱为有机碱,优选三乙胺、n,n-二异丙基乙胺、吡啶和4-二甲氨基吡啶中的一种或多种,更优选的,碱为n,n-二异丙基乙胺。在一些优选实施方式中,步骤s1中亲核取代反应的温度为70~80℃,在此温度范围内,得到的中间体iii的产率较高。在一些优选实施方式中,每克中间体iii加入7~35ml第二有机溶剂,更优选的,每克中间体iii加入8~12ml第二有机溶剂,以保证化合物iii充分溶解。在一些优选实施方式中,强碱溶液的摩尔浓度为1~10mol/l,更优选的,其摩尔浓度为2~4mol/l。在一些优选实施方式中,第二有机溶剂为四氢呋喃、二氯甲烷、甲醇、乙醇和n,n-二甲基甲酰胺中的一种或几种,更优选的,第二有机溶剂为四氢呋喃。在一些优选实施方式中,强碱溶液中的强碱为氢氧化锂、氢氧化钠和氢氧化钾中的一种或几种,更优选的,强碱为氢氧化钠。在一些优选实施方式中,步骤s2中水解反应温度为20~25℃,在此温度范围内,得到的阿哌沙班有关物质a的产率较高。上述反应的反应进程可采用本领域中的常规监测方法进行监测(例如tlc、hplc或nmr),一般以化合物ii和中间体iii消失时为反应的终点。上述反应的步骤(1)中柱层析纯化的洗脱剂由二氯甲烷和甲醇构成,二氯甲烷:甲醇的体积比为100:1。本发明的化合物ii购自苏州卡普维尔医药科技有限公司;本发明中的其他化学试剂均为市场购买所得。为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。本发明中的实验方法,如无特殊说明,均为常规方法。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。下列实施例中的液相条件为:c18键合硅胶柱(glsciencesinc.inertsilods-spc18,4.6mm×250mm,5μm等柱)为色谱柱,以去离子水为流动相a,以含体积百分比为0.1%的三氟乙酸的乙腈为流动相b,按下表进行梯度洗脱:时间(分钟)流动相a(%)流动相b(%)087133871317653525109030109030.018713358713流速为1.0ml/min,柱温为35℃,检测波长为280nm。实施例1:本发明的实施例1提供了一种中间体iii的制备方法,其合成路线如下:具体采用如下方法制备:将化合物ii(2.0g,4.9mmol)、n,n-二异丙基乙胺(1.2ml,7.4mmol)、5-氯戊酸乙酯(0.97g,5.9mmol)和n,n-二甲基甲酰胺(12ml)加入反应瓶中混合均匀,将反应液在80℃下反应7小时,tlc检测反应直至化合物ii消失,向反应液中加入水(12ml),析出固体后,继续搅拌0.5小时,抽滤,滤饼用水和异丙醇洗涤,得到粗品,粗品经柱层析纯化得到淡黄色固体,即化合物iii。采用本方法得到的淡黄色固体1.8g,收率68%。对本实施例中制得的中间体iii进行鉴定,得到如下结果:esi-ms(m/z):535.2;1hnmr(500mhz,cdcl3):δ8.02–7.80(m,1h),7.37–7.22(m,1h),7.11–7.01(m,1h),6.84–6.65(m,1h),4.31–4.15(m,2h),4.01(q,j=11.8hz,1h),3.79(s,2h),3.30(t,j=11.6hz,2h),2.32(t,j=15.0hz,1h),1.72–1.56(m,1h),1.48(m,1h),1.30(t,j=11.8hz,1h),1.07(t,j=11.8hz,1h)。实施例2:本发明的实施例2提供了一种中间体iii的制备方法,具体采用如下方法制备:将化合物ii(1.3g,3.2mmol)、三乙胺(0.7ml,4.8mmol)、5-氯戊酸乙酯(0.63g,3.8mmol)和n,n-二甲基甲酰胺(8ml)加入反应瓶中混合均匀,将反应液在80℃下反应7小时,tlc检测反应直至化合物ii消失,向反应液中加入水(8ml),析出固体后,继续搅拌0.5小时,抽滤,滤饼用水和异丙醇洗涤,得到粗品,粗品经柱层析纯化得到淡黄色固体,即化合物iii。采用本方法得到的淡黄色固体1.1g,收率64%。实施例3:本发明的实施例3提供了一种中间体iii的制备方法,具体采用如下方法制备:将化合物ii(1.5g,3.7mmol)、碳酸钾(0.77g,5.5mmol)、5-氯戊酸乙酯(0.73g,4.4mmol)和n,n-二甲基甲酰胺(10ml)加入反应瓶中混合均匀,将反应液在80℃下反应7小时,tlc检测反应直至化合物ii消失,向反应液中加入水(10ml),析出固体后,继续搅拌0.5小时,抽滤,滤饼用水和异丙醇洗涤,得到粗品,粗品经柱层析纯化得到淡黄色固体,即化合物iii。采用本方法得到的淡黄色固体1.0g,收率51%。实施例4:本发明的实施例4提供了一种化合物a的制备方法,其合成路线如下:具体采用如下方法制备:将化合物iii(1.5g,2.8mmol)和四氢呋喃(12ml)加入反应瓶中,在冰浴下向反应液中加入氢氧化钠水溶液(2ml,2mol/l),将反应液的ph调至11,加完后将反应液升至25℃反应1小时,tlc检测反应直至化合物iii消失,将反应体系温度降至0℃,用稀盐酸(2mol/l)将反应体系的ph调至4~5,析出固体后,继续搅拌1小时,抽滤,粗品在45℃真空干燥12h后,再将粗品用乙醇重结晶后得到白色固体,即化合物a。采用本方法得到的白色固体0.9g,收率67%,纯度99.1%。对本实施例中制得的化合物a进行鉴定,得到如下结果:esi-ms(m/z):479.1;1hnmr(500mhz,dmso-d6):δ12.41(s,1h),11.78(s,1h),7.95(s,2h),7.27(s,2h),7.08(s,2h),6.77(s,2h),4.28(d,j=3.5hz,2h),3.79(s,3h),3.50(s,1h),3.30(s,4h),2.21(s,2h),1.50(d,j=20.0hz,4h)。阿哌沙班粗品hplc图谱见图1,由图1可以看出,杂质含量大于1%的出峰时间为3.739;本实施例中制得的化合物a的hplc图谱见图2,由图2可知,化合物a的出峰时间为3.737;通过核磁、质谱和比对保留时间,进一步确证了化合物a的结构。实施例5:本发明的实施例5提供了一种化合物a的制备方法,具体采用如下方法制备:将化合物iii(1.5g,2.8mmol)和n,n-二甲基甲酰胺(12ml)加入反应瓶中,在冰浴下向反应液中加入氢氧化钠水溶液(2ml,2mol/l),将反应液的ph调至11,加完后将反应液升至25℃反应1小时,tlc检测反应直至化合物iii消失,将反应体系温度降至0℃,用稀盐酸(2mol/l)将体系的ph调至4~5,析出固体后,继续搅拌1小时,抽滤,粗品在45℃真空干燥12h后,再将粗品用乙醇重结晶得到白色固体,即化合物a。采用本方法得到的白色固体0.7g,收率52%,纯度98.3%。实施例6:本发明的实施例6提供了一种化合物a的制备方法,具体采用如下方法制备:将化合物iii(1.5g,2.8mmol)和n,n-二甲基甲酰胺(12ml)加入反应瓶中,在冰浴下向反应液中加入氢氧化锂水溶液(2ml,2mol/l),将反应液的ph调至11,加完后将反应液升至25℃反应1小时,tlc检测反应直至化合物iii消失,将反应体系温度降至0℃,用稀盐酸(2mol/l)将体系的ph调至4~5,有固体析出,继续搅拌1小时,抽滤,粗品在45℃真空干燥12h后,再将粗品用乙醇重结晶得到白色固体,即化合物a。采用本方法得到的白色固体0.6g,收率45%,纯度98.6%。实施例7:本发明的实施例7提供了一种化合物a的制备方法,具体采用如下方法制备:将化合物iii(1.5g,2.8mmol)和甲醇(12ml)加入反应瓶中,在冰浴下向反应液中加入氢氧化钠水溶液(2ml,2mol/l),将反应液的ph调至11,加完后将反应液升至25℃反应1小时,tlc检测反应直至化合物iii消失,将体系温度将至0℃,用稀盐酸(2mol/l)将体系的ph调至4~5,有固体析出,继续搅拌1小时,抽滤,粗品在45℃真空干燥12h后,再将粗品用乙醇重结晶得到白色固体,即化合物a。采用本方法得到的白色固体0.8g,收率60%,纯度99.0%。本发明中提供的化合物a的制备方法,得到的阿哌沙班有关物质a纯度高,可作为有关物质a的标准品使用,方便阿哌沙班制备过程中的质量控制研究,对临床用药安全监测起到了至关重要的作用。以上所述本发明的具体实施方式,并不构成对本发明保护范围的限定。任何根据本发明的技术构思所做出的各种其他相应的改变与变形,均应包含在本发明权利要求的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1