一种基于β-环糊精为核的水溶性星形聚合物及其制备方法与流程
本发明属于高分子聚合物合成技术领域,特别涉及一种基于β-环糊精为核的结构可控的水溶性星形聚合物及其制备方法。
背景技术:
β-环糊精具有内疏水外亲水的独特结构特征,其内部的疏水性空腔可以包络尺寸大小适宜的有机物分子,其外侧亲水性的羟基对高价金属离子具有螯合作用,因而具有一定的立体选择和识别性能。但由于β-环糊精在水中的溶解度仅为1.64%,当它与其他物质产生包合物后,水溶性更低,容易从溶液中结晶析出,使其应用受到极大限制。通过对β-环糊精改性并以此为核制得多种星形高分子聚合物,在药物负载、生物制药等方面有广泛应用。
现有技术中以β-环糊精为核的星形聚合物的制备方法一般是通过“先核后臂”来合成。该合成方法存在合成聚合物结构不可控,不能保证聚合物臂均一性等缺点。
技术实现要素:
本发明的目的是针对现有以β-环糊精为核的星形聚合物的制备方法存在的聚合物结构不可控,聚合物臂不均一的问题,提供一种基于β-环糊精为核的结构可控的水溶性星形聚合物及其制备方法。
为了实现上述目的和其它优点,本发明提供了一种基于β-环糊精为核的水溶性星形聚合物,该聚合物的分子结构式如下:
式中,
优选的是,该聚合物由β-环糊精、反应单体甲基丙烯酸甲酯或甲基丙烯酸二甲氨基乙酯、链转移剂二硫代酯或三硫代酯反应制成。
进一步优选的是,该聚合物由β-环糊精、甲基丙烯酸二甲氨基乙酯、链转移剂(2-((丁基硫烷基)-硫代羰基))-丙酸反应制成。聚合物的分子结构式如下:
进一步优选的是,n=21。
一种基于β-环糊精为核的水溶性星形聚合物的制备方法,包括如下步骤:
步骤s1、反应单体与链转移剂之间进行可逆加成-断裂链转移聚合(raft)反应合成结构可控且分子量分布较窄的水溶性链状聚合物,并对其链端进行巯基转化;其中。反应单体为丙烯酸酯或丙烯酰胺,链转移剂为二硫代酯或三硫代酯。
步骤s2、对β-环糊精核进行改性,引入碳碳双键;
步骤s3、通过“巯基-烯”点击化学(click)反应,将链状聚合物接枝到改性β-环糊精核上,形成以β-环糊精为核的结构可控的水溶性星形聚合物。
上述制备β-环糊精为核的水溶性星形聚合物的制备流程如下:
式中,
优选的是,当反应单体为甲基丙烯酸二甲氨基乙酯,链转移剂为(2-((丁基硫烷基)-硫代羰基))-丙酸时,所述步骤s1包括:
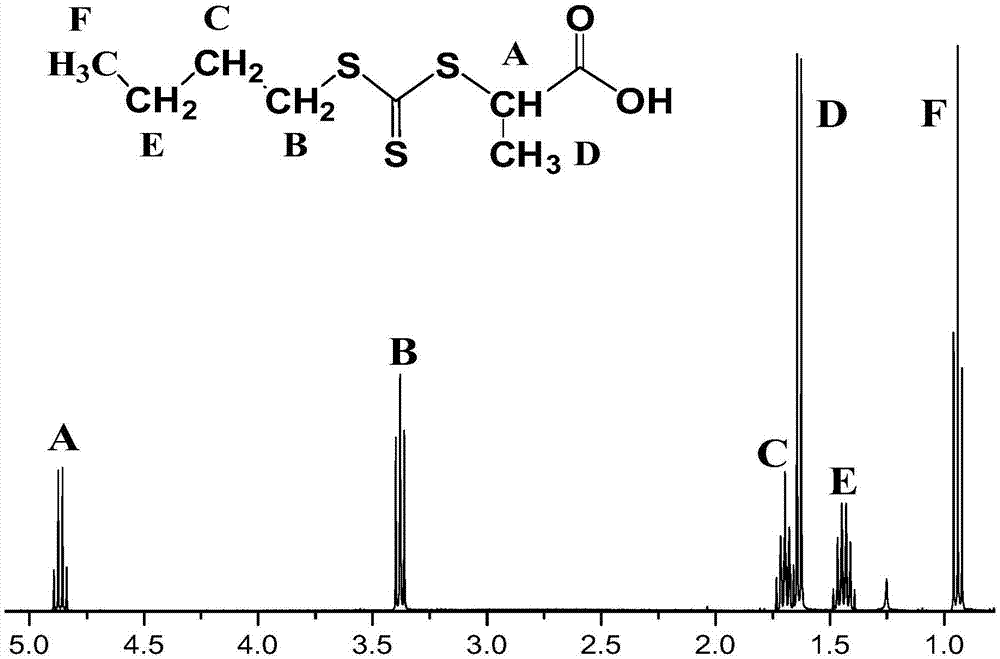
步骤s11、制备链转移剂(2-((丁基硫烷基)-硫代羰基))-丙酸:将naoh、丙酮加入到含有丁硫醇的去离子水中,室温搅拌反应0.5h,然后加入二硫化碳,在冰浴下搅拌反应0.5h后滴加2-溴丙酸和naoh,使反应体系温度低于30℃,放热反应终止后,加入去离子水,室温下搅拌反应24h,反应结束后将反应混合物用去离子水稀释,然后加入到冰的hcl溶液中得到黄色油状物,继续保持在冰浴中直至油固化并过滤分离固体,用冷水洗涤,干燥,将产物从正己烷中轻轻搅拌重结晶,得到亮黄色固体结晶产物。丁硫醇、二硫化碳、2-溴丙酸摩尔比为:1:1.155:1.025。
步骤s12、将甲基丙烯酸二甲氨基乙酯dmaema、偶氮二异丁腈aibn、步骤s11制备的链转移剂溶于溶剂1,4-二氧六环中,氩气氛围下70℃恒温油浴中反应8h,得到pdmaema链段;其中,dmaema、aibn、链转移剂的摩尔比为1:0.012:0.063。
步骤s13、将pdmaema、偶氮二异丁腈aibn、丙烯酸叔丁酯t-ba溶于溶剂1,4-二氧六环中,氩气氛围下80℃恒温油浴中反应8h,得到pdmaema-ptba链段;其中,pdmaema、aibn、t-ba的摩尔比为1:0.0075:7.8。
步骤s14、将pdmaema-ptba链段溶于有机溶剂中,加入水合肼,室温下搅拌反应1h,得到链状聚合物巯基化pdmaema-ptba。其中,pdmaema-ptba和水合肼的摩尔比为1:5。
所述步骤s2具体为:将β-环糊精溶于n,n-二甲基甲酰胺溶剂中,然后加入适量naoh,室温搅拌反应1h,然后滴加3-溴丙烯,5-10℃低温下剧烈搅拌5天,分离,得到改性的21臂β-环糊精。其中,β-环糊精、naoh、3-溴丙烯的摩尔比为1:82.62:86.92。
所述步骤s3包括:
步骤s31、将巯基化pdmaema-ptba、改性21臂β-环糊精和光引发剂溶于n,n-二甲基甲酰胺溶剂中,氩气氛围下室温紫外光照射反应24h,透析分离,得到星形聚合物pdmaema-ptba;其中,pdmaema-ptba和21臂β-环糊精核摩尔比为1:0.035。
步骤s32、将星形聚合物pdmaema-ptba溶于二氯甲烷溶剂中,冰浴下滴加三氟乙酸,然后在冷凝回流下将体系置于室温下搅拌反应24h,分离出白色不溶物,得到星形聚合物,即聚甲基丙烯酸二甲氨基乙酯-聚丙烯酸pdmaema-paa。
聚甲基丙烯酸二甲氨基乙酯-聚丙烯酸pdmaema-paa的制备反应流程如下:
与现有技术相比,本发明具有以下有益效果:
本发明采用“先臂后核”的合成方法合成以β-环糊精为核的星形聚合物,该合成方法能够得到结构可控且明确的水溶性星形聚合物。与现有的“先核后臂”合成方法相比,本发明的合成的聚合物结构能更加的明确,首先在星形聚合物的聚合物臂与核接枝之前,每条聚合物臂在“接核”前都已准确表征,保证了聚合物臂的均一性,其次通过控制聚合物臂与核的比例,可以制备不同臂数的星形聚合物,从而达到臂数可控的效果。
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
附图说明
图1、链转移剂(2-((丁基硫烷基)-硫代羰基))-丙酸的核磁氢谱。
图2、pdmaema链段的核磁氢谱。
图3、pdmaema-ptba链段的核磁氢谱。
图4、21臂β-环糊精核的核磁氢谱。
图5、星形聚合物pdmaema-paa的核磁氢谱。
具体实施方式
下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
应当理解,本文所使用的诸如“具有”、“包含”以及“包括”术语并不配出一个或多个其它元件或其组合的存在或添加。
一种制备聚甲基丙烯酸二甲氨基乙酯-聚丙烯酸pdmaema-paa的方法,选用甲基丙烯酸二甲氨基乙酯为反应单体,(2-((丁基硫烷基)-硫代羰基))-丙酸为链转移剂,具体制备操作步骤如下:
步骤s1、反应单体与链转移剂之间进行可逆加成-断裂链转移聚合(raft)反应合成结构可控且分子量分布较窄的水溶性链状聚合物,并对其链端进行巯基转化。具体包括:
步骤s11、制备链转移剂(2-((丁基硫烷基)-硫代羰基))-丙酸
取质量百分浓度50%的naoh水溶液16g加入到含有18g丁硫醇的30ml去离子水中,然后加入10ml丙酮,得到无色溶液,将溶液在室温下搅拌0.5h;搅拌结束后加入14ml二硫化碳,得到澄清的橙色溶液,然后在冰浴条件下将反应混合液搅拌0.5h;结束后滴加31.4g的2-溴丙酸,再加入质量百分浓度50%的naoh水溶液16g,并使反应体系温度保持在30℃以下,放热反应终止后,加入30ml去离子水,室温下搅拌反应混合物24h;反应结束后向反应液体系中加入50ml去离子水进行稀释,然后将溶液体系加入到60ml冰的浓度10mol/l的hcl溶液中得到黄色油状物,继续保持在冰浴中直至油固化并过滤分离固体,用冷水洗涤,干燥,最后将产物从正己烷中轻轻搅拌重结晶,得到亮黄色固体结晶产物链转移剂(2-((丁基硫烷基)-硫代羰基))-丙酸。图1是链转移剂(2-((丁基硫烷基)-硫代羰基))-丙酸的核磁氢谱,对其进行分析可以得到目标链转移剂已经成功合成,可以用于raft反应合成聚合物。
步骤s12、pdmaema链段的制备
将190mg链转移剂、25mg偶氮二异丁腈aibn、2g甲基丙烯酸二甲氨基乙酯dmaema和3ml1,4-二氧六环依次加入反应管中抽真空20min,并反复抽真空-通氩气处理三次,然后置于70℃恒温油浴中反应8h;反应结束后将反应体系暴露于空气中,并用适量四氢呋喃thf对聚合物进行稀释,然后倒入2kd透析袋中透析3天,最后将透析得到的溶液冻干即得到pdmaema。图2是pdmaema链段的核磁氢谱,对其进行分析可以得到pdmaema均聚物已经成功合成,且提纯较为彻底。
步骤s13、pdmaema-ptba链段的制备
将1.0g的pdmaema、2.5mg的aibn、0.2g丙烯酸叔丁酯t-ba和3.5ml的1,4-二氧六环依次加入到反应管中抽真空20min,并反复抽真空-通氩气处理三次,然后置于80℃恒温油浴中反应8h;反应结束后将反应体系暴露于空气中,并用适量四氢呋喃稀释聚合物,然后倒入2kd透析袋中透析3天,最后将透析得到的白色乳状液冻干,即得到pdmaema-ptba链段。图3是pdmaema-ptba链段的核磁氢谱,对其进行分析可以得到嵌段聚合物pdmaema-ptba成功合成,且两者的嵌段比为5:1。
步骤s14、链状聚合物巯基化pdmaema-ptba的制备
将1.0g的pdmaema-ptba链段加入到反应管中,然后倒入干燥的50ml的dmf使其充分溶解后,加入100μl的水合肼,搅拌反应1h,反应体系由原来的亮黄色变为无色透明,然后倒入2kd透析袋中透析2天,最后将透析得到的白色乳状液冻干即得到链状聚合物巯基化pdmaema-ptba。
步骤s2、对β-环糊精核进行改性,引入碳碳双键,制备21臂β-环糊精核。
将2.8156g的β-环糊精加入70ml干燥的dmf中,搅拌使β-环糊精充分溶解后加入8.2271g固体naoh,室温搅拌1h,然后低温下缓慢滴加18.50ml的3-溴丙烯,5℃下剧烈搅拌5天;反应完毕后过滤,减压浓缩,残余物用氯仿萃取3次,每次氯仿用量20ml,萃取液用去离子水洗涤,无水硫酸钠干燥过夜,最后蒸去溶剂得到浅黄色粘稠液体,即改性的21臂β-环糊精核。图4是21臂β-环糊精核的核磁氢谱,对其进行分析得到β-环糊精的21个羟基已经全部改性成功,可以用于星形聚合物的核。
步骤s3、通过“巯基-烯”点击化学(click)反应,将链状聚合物接枝到改性后的β-环糊精核上,形成以β-环糊精为核的结构可控的水溶性星形聚合物。具体包括步骤:
步骤s31、星形聚合物pdmaema-ptba的制备
将0.013g的21臂β-环糊精,1.0g的巯基化pdmaema-ptba,0.01g光引发剂184和20ml的dmf依次加入到反应管中抽真空20min,并反复抽真空-通氩气处理三次,然后在紫外光照下(λ=365nm)室温反应24h;反应结束后将反应体系倒入35kd透析袋中透析3天,最后将透析得到的溶液冻干,即得到星形聚合物pdmaema-ptba。
步骤s32、星形聚合物pdmaema-paa的制备
将1g聚合物pdmaema-ptba加入到50ml圆底烧瓶中,然后加入20ml二氯甲烷将聚合物溶解,溶解完毕后将圆底烧瓶置于冰浴中并缓慢加入0.5ml三氟乙酸,然后在冷凝回流下将体系置于室温下搅拌反应24h;反应结束后将生成的白色不溶物分离出,然后溶于适量去离子水中,并倒入35kd透析袋中透析3天;透析干净后将溶液在冻干机中冻干,即得到水解星形嵌段聚合物pdmaema-paa。图5是星形聚合物pdmaema-paa的核磁氢谱,对其进行分析得到pt-ba段水解为paa,星形聚合物pdmaema-paa成功合成。
综上所述,本发明提供了一种基于β-环糊精为核的结构可控的水溶性星形聚合物的制备方法。通过“先臂后核”法制备,主要利用可逆加成-断裂链转移聚合(raft)与“巯基-烯”点击化学反应相结合的方法制备。可逆加成-断裂链转移聚合(raft)作为一种可控/活性自由基聚合方法,具有适用单体范围广且后处理简单等特点,而“巯基-烯”点击化学反应具有条件温和、反应速率快等特点,将两者结合在聚合物功能分子设计尤其星形聚合物中发挥着越来越强大的作用。与现有的“先核后臂”合成方法相比,本发明的合成的聚合物结构能更加的明确,每条聚合物臂在“接核”前都已准确表征,保证了聚合物臂的均一性。
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用。它完全可以被适用于各种适合本发明的领域。对于熟悉本领域的人员而言,可容易地实现另外的修改。因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
- 还没有人留言评论。精彩留言会获得点赞!