一种制备3-羟基丙醛的方法及制备1,3-丙二醇的方法与流程
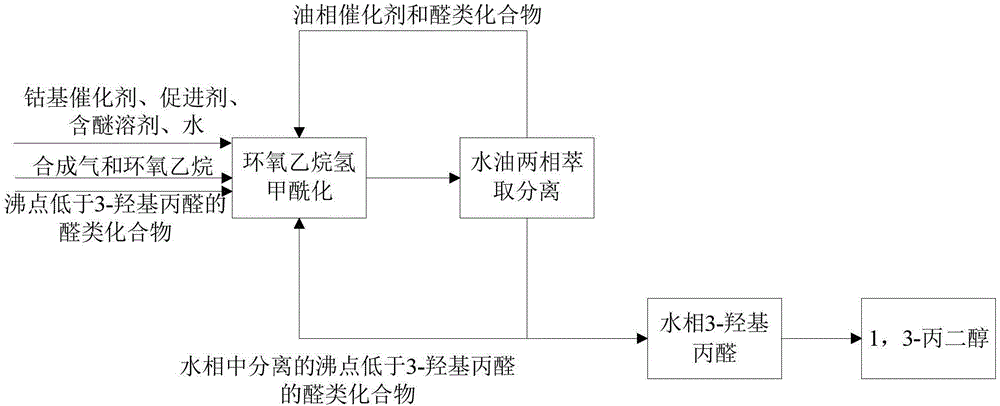
本发明涉及一种环氧乙烷氢甲酰化制备3-羟基丙醛的方法及制备1,3-丙二醇的方法。
背景技术:
环氧乙烷氢甲酰化制备3-羟基丙醛在j.fable的用一氧化碳新合成物(newsystheseswithcarbonmonoxide)(1980)第131-132页中已有描述。该反应是用钴基催化剂或膦改性的钴基催化剂催化的。3-羟基丙醛可通过加氢获得1,3-丙二醇(pdo),pdo是生产纤维和薄膜用聚酯中间体。
壳牌公司进一步开发了钴基催化剂氢甲酰化合成3-羟基丙醛的工艺,如专利us5256827和us5563302,使用季磷或含磷的氧化物时,反应的活性和选择性均不高,且有较多的副产物乙醛生成。wo96/10550,wo97/33851所描述,其中钴催化剂通过水油两相萃取分离,使得绝大部分钴催化剂溶解在油相如甲基叔丁基醚相中返回反应器,而产品3-羟基丙醛从水相分离得到,由于萃取分离,使得油相中不可避免的混有一定的水,而反应体系中过量的水存在,3-羟基丙醛和1,3-丙二醇的选择性降低到可接受的水平以下,专利提到最佳水含量为1-2.5%,然而即使在1-2.5%的水含量,研究发现与不含水的反应体系相比,其3-羟基丙醛的选择性仍然有明显的降低。
因而需要开发一种制备3-羟基丙醛的方法,在含有一定水含量的反应体系中,仍能保持高的3-羟基丙醛的选择性,并尽量减少副产物的生成。
技术实现要素:
本发明所要解决的技术问题是现有技术中主产物3-羟基丙醛选择性低、反应活性低,本发明提供一种环氧乙烷氢甲酰化制备3-羟基丙醛的方法及制备1,3-丙二醇的方法,本发明在氢甲酰化反应中,通过往反应体系中添加额外的沸点低于3-羟基丙醛的醛类化合物,或者把反应体系中副产的醛类化合物重新添加到反应系统中,使得反应体系中即使维持一定的水浓度和较高的反应活性下,仍能保持高的3-羟基丙醛选择性,或者把反应中副产生成的乙醛、正丙醛、丙醛等醛类化合物经过分离送回到反应釜或者通过添加相应的醛类来提升反应体系中其它醛类化合物的浓度来抑制副反应,实现高产率和高选择性生成3-羟基丙醛的生产。在反应体系中通过添加一定量的醛类化合物,并保持一定的水含量,来提升反应活性和反应主产物的选择性。
为实现上述目的及其他相关目的,本发明第一方面提供一种制备3-羟基丙醛的方法,在制备3-羟基丙醛时加入醛类化合物,所述醛类化合物的沸点低于3-羟基丙醛。
加入沸点低于3-羟基丙醛的醛类化合物有利于额外的醛类化合物与产品3-羟基丙醛的分离与重复添加使用。
优选地,在通过环氧乙烷与合成气制备3-羟基丙醛时加入醛类化合物,所述醛类化合物的沸点低于3-羟基丙醛。
优选地,在钴基催化剂、促进剂、含醚溶剂、水和醛类化合物存在下,环氧乙烷与合成气进行氢甲酰化反应制备得到3-羟基丙醛。
钴基催化剂可以是改性的羰基钴化合物,例如膦配位体的化合物或未改性的羰基钴化合物。虽然膦配位体的催化剂对该氢甲酰化反应有些活泼,但由于膦配位体太昂贵,希望使用一种方法,其中不使用膦配位体便可完成良好的产率和选择性,因此未改性的羰基钴化合物是优选的。钴基催化剂可加到氢甲酰化中以羰基钴的形式如八羰基二钴或者氢化羰基钴。也能以其它形式包括金属、带载体金属、氢氧化物、氧化物、碳酸盐、硫酸盐、乙酰丙酮化物、脂肪酸盐或钴盐水溶液加入,但应调整操作条件以形成羰基钴。例如可以通过氢气和一氧化碳反应,反应条件包括至少50℃的温度,0.8mpa的一氧化碳分压。为了更快地反应,需要一定的八羰基二钴母液,温度在120-180℃,一氧化碳分压至少为4.0mpa,且有一定的氢气分压。其中钴基催化剂的量基于反应混合物重量,为0.01wt%到1.0wt%。催化剂优选存在于一氧化碳的氛围中,该氛围可有效防止催化剂暴露在氧气或空气中。最经济和优选的催化剂活化和再生的方法是在氢甲酰化氛围中,在120-150℃的温度下,将二价钴离子转化成羰基钴。
促进剂不仅加速反应,增加产物选择性,还促进钴基催化剂留在有机相中。
含醚溶剂选自甲基叔丁基醚、乙基叔丁基醚、苯基异丁基醚、乙氧基乙基醚、二苯醚和二异丙醚中的至少一种。
优选地,上述方法还包括如下技术特征中的至少一项:
1)所述醛类化合物的沸点低于100℃;
2)醛类化合物与含醚溶剂的质量比为1~20:100,如1~3:100、3~5:100、3~6.6:100、6.6~8:100、8~18.6:100或18.6~200:100;更优选地,醛类化合物与含醚溶剂的质量比为3.0~10.0:100;进一步更优选地,醛类化合物与含醚溶剂的质量比为5.0~8.0:100;
3)反应体系中间歇加入或连续加入醛类化合物;
4)所述醛类化合物为外加或者来自制备3-羟基丙醛时副产分离的醛类化合物,如乙醛、丙醛和丙烯醛及其混合物。
更优选地,特征1)中,所述醛类化合物选自甲醛、乙醛、乙二醛、丙醛、丙烯醛、正丁醛、异丁醛、正戊醛和2-甲基戊醛中的至少一种。进一步更优选地,所述醛类化合物选自甲醛、乙醛、丙醛、丙烯醛、正丁醛、异丁醛和2-甲基戊醛中的至少一种。
更优选地,水与含醚溶剂的质量比为0.2~8:100,如0.2~2:100、2~2.5:100、2.5~2.8:100、2.8~3:100、3~4.3:100或4.3~8:100。进一步更优选地,水与含醚溶剂的质量比为1.5~5.0:100;再进一步更优选地,水与含醚溶剂的质量比为2.6~3.5:100。
更优选地,水为外加或者来自钴基催化剂油水萃取分离中带入反应体系的水。
更优选地,反应体系中,钴基催化剂与含醚溶剂的质量比为0.1~1.0:100,如0.1~0.24:100、0.24~0.27:100、0.27~0.67:100或0.67~1.0:100。进一步更优选地,钴基催化剂与含醚溶剂的质量比为0.05~0.40:100。
更优选地,所述促进剂为第ⅴ族阳离子的亲油性季盐。
进一步更优选地,所述第ⅴ族阳离子的亲油性季盐选自二甲基十二胺、壬基吡啶和三苯基氧膦中的至少一种。
更优选地,促进剂与钴基催化剂中钴的摩尔比为0.1~2.0:1,如0.1~0.4:1、0.4~0.6:1、0.6~0.7:1、0.7~0.8:1或0.8~2.0:1。
环氧乙烷与合成气进行氢甲酰化反应可采用现有技术中的工艺条件,例如:1)合成气h2:co的分压比为5:1~1:5;优选地,合成气中h2:co的分压比为3:1~1:1;2)氢甲酰化反应温度小于100℃;优选地,氢甲酰化反应温度为70~90℃;3)氢甲酰化反应压力为5.0~15mpa;优选地,氢甲酰化反应压力为8.0~12mpa;4)氢甲酰化反应后混合物中的3-羟基丙醛的浓度小于15wt%;优选地,氢甲酰化反应后混合物中的3-羟基丙醛的浓度为5~10wt%。
本发明第二方面提供一种制备1,3-丙二醇的方法,包括如下步骤:
1)如上述任一项所述的方法获得3-羟基丙醛;
2)将步骤1)得到的3-羟基丙醛加氢获得1,3-丙二醇。
本发明在反应体系中加入沸点低于3-羟基丙醛的醛类化合物,意外地发现,反应的主产物3-hpa的选择性提高。当添加沸点低于3-羟基丙醛的醛类化合物,虽然通过气相色谱无法观察到沸点低于3-羟基丙醛的醛类化合物和3-hpa的相互作用,但通过核磁碳谱研究发现,当添加沸点低于3-羟基丙醛的醛类化合物时,如丙醛、丁醛、戊醛,其它醛类化合物与3-hpa存在相互作用,可以形成六元环的化合物,有利于降低体系中游离的3-hpa和水合的3-hpa的含量,使得环氧乙烷反应平衡朝3-hpa移动。
如通过核磁碳谱研究发现,当添加额外的乙醛时,乙醛与3-hpa形成的六元环的物质的量高于3-hpa的二聚体,有利于降低体系中游离的3-hpa和水合的3-hpa的含量,使得环氧乙烷反应平衡朝3-hpa移动;同时额外的乙醛的加入,也有利于增加体系中游离和水合的乙醛的含量,抑制环氧乙烷朝乙醛副产物生成。
本发明的制备3-羟基丙醛的方法可以让反应体系中保持一定的水含量,从而提高反应的活性,同时仍然能保持较高的3-hpa的选择性,使得反应工艺更加具有可操作性,工艺经济性更好。
附图说明
图1为本发明制备3-羟基丙醛的方法的流程图。
具体实施方式
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
制备3-羟基丙醛可以间歇进行也可以连续进行。
在间歇的反应过程中,在一定体积的高压反应釜中,称量一定量的钴催化剂前体、促进剂,溶解到含一定水含量的醚类溶剂中,添加一定浓度的沸点低于3-羟基丙醛的醛类化合物,充入一定比例的合成气,使得h2/co分压保持一定比例,然后加入环氧乙烷,在一定的温度和压力条件下反应3h,通过稳定工况下合成气的吸收量来计算反应的时空收率(sty):每小时每升反应溶液生成的3-羟基丙醛的摩尔数,通过色谱分析反应后的产物组成,来计算环氧乙烷转化成3-羟基丙醛的选择性。
在连续的反应过程中,在一定体积的高压反应釜中,添加一定浓度沸点低于3-羟基丙醛的额外的醛类化合物,连续通入环氧乙烷和合成气,在钴催化剂和促进剂的作用下,使环氧乙烷氢甲酰化生成3-羟基丙醛,生成的产物3-羟基丙醛通过水相萃取分离得到,进一步加氢生成1,3-丙二醇。从水相分离得到的低沸点醛类化合物循环回反应器,并保持一定浓度的低沸点醛类化合物,来提升在一定水含量存在下环氧乙烷氢甲酰化合成3-羟基丙醛的选择性。
在实施本发明方法时,可获得提高3-羟基丙醛的产率提高20%~35%。
对比例1
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.5mol·h-1·l-1,eo的转化率在95%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在60%。
对比例2
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.55mol·h-1·l-1,eo的转化率在98%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在50%。
对比例1不添加水,反应的时空收率相对较低,反应产物3-羟基丙醛的选择性较好;对比例2加入7.5克的水,反应体系中的水含量约2.5%,反应的时空收率相对较高,但产物3-羟基丙醛的选择性明显降低。
实施例1
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克乙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.62mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在90%。
实施例2
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入24g水,额外添加60克乙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在98%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在87%。
实施例3
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入0.6g水,额外添加3克乙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在98%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在78%。
实施例4
在1升的高压反应釜中,添加八羰基二钴3g,n,n-二甲基十二胺0.375g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克乙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.61mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在80%。
实施例5
在1升的高压反应釜中,添加八羰基二钴0.3g,n,n-二甲基十二胺0.75g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克乙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.58mol·h-1·l-1,eo的转化率在98%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在81%。
实施例6
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克正丙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.61mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在89%。
实施例7
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加9克丙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在85%。
实施例8
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加12克乙醛和12克丙醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.61mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在88%。
实施例9
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,乙基叔丁基醚300g,加入9.0g水,额外添加15克正丁醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在85%。
实施例10
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入6.0g水,额外添加15克2-甲基戊醛,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在85%。
实施例11
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,苯基异丁基醚300g,加入7.5g水,额外添加15克甲醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.58mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在80%。
实施例12
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,乙氧基乙基醚300g,加入7.5g水,额外添加15克异丁醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.58mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在81%。
实施例13
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克丙烯醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在73%。
实施例14
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,二苯醚300g,加入7.5g水,额外添加15克乙二醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在81%。
实施例15
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,二异丙醚300g,加入7.5g水,额外添加15克正戊醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在78%。
实施例16
在1升的高压反应釜中,添加八羰基二钴2.0g,n,n-二甲基十二胺2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克2-甲基戊醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在76%。
实施例17
在1升的高压反应釜中,添加八羰基二钴2.0g,壬基吡啶1.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克乙醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.62mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在85%。
实施例18
在1升的高压反应釜中,添加八羰基二钴2.0g,三苯基氧膦2.0g,甲基叔丁基醚mtbe300g,加入7.5g水,额外添加15克乙醛溶液,氢气置换三次,用氢气充压至2.8mpa,然后用1:1的合成气补充到8.0mpa,再搅拌,升温加热到80℃时,再补充1:1的合成气至10.0mpa,使得h2/co=1.8,恒温半小时后,再以0.5ml/min的速率加入环氧乙烷27g,反应开始后用质量流量计计量瞬时流量和累积流量,反应持续3.0h,反应结束后进行色谱分析。通过稳定工况下的吸收流量估算反应的时空收率在0.61mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的选择性在86%。
对比例3
在连续的反应工艺中,在5l的反应器中,在80℃和10.0mpa的条件下,连续加入环氧乙烷,并通入一定量的合成气,以维持反应压力10.0mpa不变,并控制气相h2/co的分压比例在2.0,反应体系中溶剂为甲基叔丁基醚,钴浓度维持在2200ppm,n,n-二甲基十二胺在0.56%,由于产品3-羟基丙醛分离需要经过水萃取分离,反应体系中会引入一定的水含量,水含量维持在2.5-3.5%之间,连续工艺中,不额外添加其它醛类化合物,也不把反应副产的低沸点醛分离返回反应器,反应连续运行200h,平均时空收率0.55mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的反应选择性在65%。
实施例19
在连续的反应工艺中,在5l的反应器中,在80℃和10.0mpa的条件下,连续加入环氧乙烷,并通入一定量的合成气,以维持反应压力10.0mpa不变,并控制气相h2/co的分压比例在2.0,反应体系中溶剂为甲基叔丁基醚,钴质量浓度维持在2200ppm,n,n-二甲基十二胺为0.56wt%,由于产品3-羟基丙醛分离需要经过水萃取分离,反应体系中会引入一定的水含量,水含量维持在2.5-3.5wt%之间,连续工艺中,添加乙醛和丙醛化合物,或者把反应副产的低沸点醛乙醛和丙醛分离返回反应器,使得反应器系统内乙醛和丙醛的总含量在6-15wt%,反应连续运行200h,平均时空收率0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的反应选择性在85%。
实施例20
在连续的反应工艺中,在5l的反应器中,在80℃和10.0mpa的条件下,连续加入环氧乙烷,并通入一定量的合成气,以维持反应压力10.0mpa不变,并控制气相h2/co的分压比例在2.0,反应体系中溶剂为甲基叔丁基醚,钴质量浓度维持在2200ppm,n,n-二甲基十二胺为0.56wt%,由于产品3-羟基丙醛分离需要经过水萃取分离,反应体系中会引入一定的水含量,水含量维持在2.5-3.5wt%之间,连续工艺中,添加5%的丁醛,或者把反应副产的低沸点醛乙醛和丙醛分离返回反应器,使得反应器系统内丁醛和其它低沸点醛的总含量在6-15wt%,反应连续运行200h,平均时空收率0.6mol·h-1·l-1,eo的转化率在99%以上,根据气相色谱分析数据,产物3-羟基丙醛的反应选择性在86%。
实施例21
把实施例2反应结束后的油相样品用水萃取,水相样品进行核磁分析,通过核磁分析,水相中的产品,最主要的存在形式为:乙醛和3-hpa缩聚的产物
虽然通过气相色谱分析,只观察到乙醛和3-hpa的存在形式,但通过核磁观察到了醛与醛之间的相互作用。由于额外低沸点醛的加入如乙醛的加入,使得乙醛与3-hpa形成的六元环的物质的量明显高于3-hpa的二聚体、游离的3-hpa和水合的3-hpa的含量,使得环氧乙醛反应平衡有利于朝3-hpa移动;同时额外的乙醛的加入,也有利于增加体系中游离和水合的乙醛的含量,抑制环氧乙烷朝乙醛副产物生成。
实施例22
把实施例6反应结束后的油相样品用水萃取,水相样品进行核磁分析,通过核磁分析,水相中的产品,最主要的存在形式为:丙醛和3-hpa缩聚的产物
虽然通过气相色谱分析,只观察到丙醛和3-hpa的存在形式,但通过核磁观察到了醛与醛之间的相互作用。由于额外低沸点醛的加入如丙醛的加入,使得丙醛与3-hpa形成的六元环的物化合物,使得环氧乙醛反应平衡有利于朝3-hpa移动;同时额外的丙醛的加入,也有利于增加体系中游离和水合的丙醛醛的含量,抑制丙醛副产物生成。
以上所述,仅为本发明的较佳实施例,并非对本发明任何形式上和实质上的限制,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还将可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。凡熟悉本专业的技术人员,在不脱离本发明的精神和范围的情况下,当可利用以上所揭示的技术内容而做出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对上述实施例所作的任何等同变化的更动、修饰与演变,均仍属于本发明的技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!