一种立构复合体聚乳酸的制备方法与流程
本发明涉及生物医学材料领域,尤其是涉及一种立构复合体聚乳酸的制备方法。
背景技术:
当前使用的聚乳酸具有脆性大、韧性低、疏水性强和细胞粘附差等缺陷,导致其应用受限。聚乳酸的脆性大导致制备的支架材料强度差,致使手术操作性不便。其不可控的降解性能会造成在降解过程中局部过多的乳酸浓度太高,引起较严重的炎性反应,是限制其生物医学应用的主要因素。材料化学组成决定了材料的结构,而材料的结构对材料性能有着重要影响。同时,高分子量聚乳酸及其共聚物的可控合成仍然是当前的一大挑战。
对聚乳酸来说,不同手性结构的乳酸经聚合后会具有不同的构象结构,左旋丙交酯(l-la)和右旋丙交酯(d-la)的开环聚合会得到左旋聚乳酸(plla)和右旋聚乳酸(pdla)。其中等量l-la和d-la的混合物构成外消旋体(dlla),经开环聚合后将获得外消旋聚乳酸(pdlla)。左旋聚乳酸(plla)具有较高的规整度,因此呈现较好的结晶性能。plla良好的结晶性能赋予其较高的强度和耐热性能,但脆性较大,降解性能不可控。与plla相比,pdlla具有均一的非晶结构,因此展现出更好的韧性和降解可控性,然而其强度和加工性能方面不如前者。
专利cn106810677a将含有游离酸的丙交酯、噁唑啉类化合物与异氰酸酯类化合物混合,在锡类催化剂的作用下进行聚合反应,得到高分子量聚乳酸。专利cn101186687b以丙交酯为原料,在丙交酯中加入亚磷酸二烃基酯后在减压或惰性气体保护条件下使其发生开环聚合反应,得到聚乳酸。专利cn105348499b将熔融的l-丙交酯与复合催化剂体系在由波纹板型静态混合器组成的环式静态混合反应装置内充分混合后,升高反应装置温度进行初步开环聚合反应,再进一步升高温度制l-聚乳酸。专利cn106700040a以双螺杆挤出机为反应器来实施l-丙交酯的开环聚合制备聚乳酸,提高了聚乳酸的光学纯度,数均分子量和耐热性数均分子量,获得高分子量的聚乳酸产品。专利cn105001403a将提纯后的l-丙交酯和引发剂进行开环聚合生成三臂支化左旋聚乳酸预聚物,再将活化后的预聚物与d-丙交酯进行开环聚合,通过改变d-丙交酯的用量,合成不同分子量的三臂支化左旋聚乳酸-右旋聚乳酸嵌段共聚物。专利cn101580582b在惰性气氛中将l-丙交酯与引发剂一起密封于聚合反应容器中,使反应物熔融并微波聚合,制得高分子量的聚乳酸。
上述合成方法大多只能合成单一旋光度的聚乳酸,或者不能够对混旋聚乳酸的力学性能及降解性进行调控。研究人员试图通过将plla和pdlla物理共混的方式改进各自的缺点,得到了更高的晶体稳定性、熔点(约比plla高50℃)以及良好的热、力学、降解等性能,然而其通常都不是在最优条件下形成,而是伴随着两者的单独结晶,使得相容性较差,易导致相分离,影响聚合物共混物性能的改善效果,目前,不同旋光度聚乳酸物理共混的实际应用仍处于探索阶段,主要是作为pla的成核剂和流变改性剂。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种构复合体聚乳酸的制备方法,具体是使用不同比例的外消旋丙交酯和左消旋丙交酯进行共聚反应合成聚乳酸的方法,可以得到具有较好结构均一性和稳定性、及降解可控性的生物医用级聚乳酸材料,对具有可控性能的生物医用级聚乳酸的开发具有较重要的借鉴意义。
本发明的目的可以通过以下技术方案来实现:一种立构复合体聚乳酸的制备方法,将一定比例的左旋丙交酯(lla)和外消旋丙交酯(dlla)混合,在催化剂作用下加热反应,经提纯得到聚乳酸。
包括以下步骤,
(1)将左旋丙交酯和外消旋丙交酯两种单体均匀混合,并在真空条件下进行预干燥,得到配制好的反应单体混合物;
(2)将步骤(1)中所得到的配制好的反应单体混合物中加入锡类催化剂,快速升温至反应温度,在反应温度下发生开环共聚反应,反应式如下:
反应结束后得到反应后的聚合物粗产品;
(3)将步骤(2)中所得到的聚合物粗产品进行提纯处理,之后经过干燥得到立构复合体聚乳酸成品。
进一步地,步骤(1)中左旋丙交酯和外消旋丙交酯这两种单体的质量比为5~95:95~5,其中外消旋丙交酯由等摩尔量的左旋丙交酯和右旋丙交酯配制而成。
进一步地,步骤(1)中左旋丙交酯和外消旋丙交酯这两种单体的质量比为20~80:80~20。
进一步地,步骤(1)中预干燥的温度为70℃,干燥时间为1h,以此去除聚合单体中吸附的水分。
步骤(2)中所述的锡类催化剂与反应单体混合物的质量比为0.5~5.0:5000,反应单体指左旋丙交酯和外消旋丙交酯。
进一步地,步骤(2)中所述的锡类催化剂为辛酸亚锡。
进一步地,步骤(2)中的反应温度为130℃~170℃,开环共聚反应时间为3~9h,聚合反应采用间歇式搅拌反应器,可采用油浴加热、电加热和高压蒸汽加热的方式来提供热源。
步骤(3)中所述的提纯处理为:将聚合物粗产品依次加入二氯甲烷和无水乙醚,进行2~3次的溶解沉淀提纯。即将聚合物粗产品先加入二氯甲烷中溶解,将可溶性杂质溶解,过滤不溶性杂质后,再将溶液倒入无水乙醚中将聚合物沉淀出来,重复2~3次。
进一步地,步骤(3)中所述的立构复合体聚乳酸成品的重均分子量为181.37kda~623.05kda,数均分子量为118.96da~295.14da,聚合物分散性指数为1.52~2.11。
与现有技术相比,本发明首次提出合成混旋丙交酯的方法,通过更改不同旋光度的丙交酯的混合比例,利用一步开环聚合法构建具有不同立体构型的聚乳酸复合体,即立构复合体聚乳酸。这种聚合方式步骤简单易行,可以克服简单共混导致的相分离现象,且能控制聚乳酸的结晶度,晶粒大小以达到控制聚乳酸力学性能和降解性的目的,从而进一步提高聚乳酸的分子量,扩宽其改性范围,构建了具有较好结构均一性、稳定性和降解可控性的生物医用级聚乳酸材料。
本发明制备的生物医用级聚乳酸材料具有较好结构均一性和稳定性、及降解可控性,对具有可控性能的生物医用级聚乳酸的开发具有较重要的借鉴意义。
附图说明
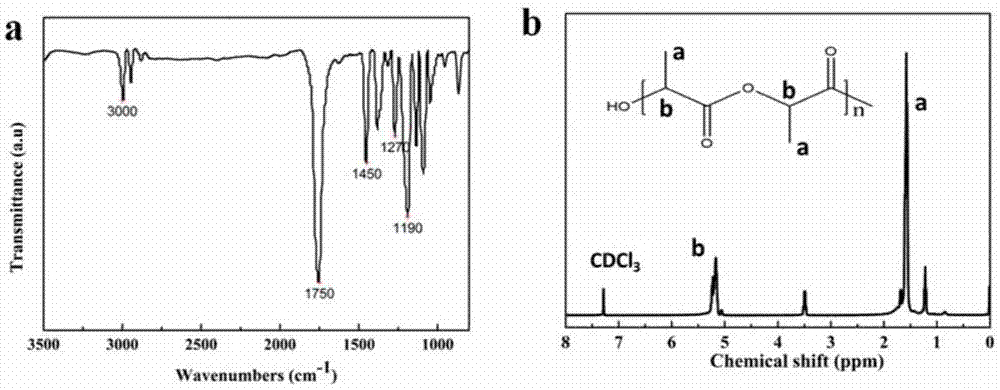
图1为本发明中所得的立构复合体聚乳酸的红外光谱图与氢-核磁谱图;
图2为本发明中所得的立构复合体聚乳酸的分子量及其分布图;
图3为本发明中所得的立构复合体聚乳酸的应力应变曲线图。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明。
下述实施例中所涉及的材料、试剂、仪器等,若无特别说明均为现有可通过正规商业途径获得的材料、试剂、仪器。下述实施例中所涉及的实验方法,检测方法等,若无特别说明,均为现有的常规实验方法,检测方法等
实施例1
立抅复合型丙交酯共聚合成聚乳酸
将比例为30:70的左旋丙交酯(lla)和外消旋丙交酯(dlla)装入施兰克瓶中,加入适量催化剂—辛酸亚锡(sn(oct)2),在真空干燥和磁力搅拌将原料于70℃下预干燥1小时。快速升温到130-170℃,并在该温度下反应3~9h。产物经二氯甲烷和无水乙醚2-3次溶解沉淀提纯处理,真空干燥后得到聚乳酸样品,标记为pdlla,放置于-20℃冰箱中保存待用。得到的产物数均分子量(mn)为295.14kda,重均分子量(mw)为623.05kda,聚合物分散性指数(pdi)为2.11,转化率69.11%。
实施例2
立抅复合型丙交酯共聚合成聚乳酸
将比例为50:50的左旋丙交酯(lla)和外消旋丙交酯(dlla)装入施兰克瓶中,加入适量催化剂—辛酸亚锡(sn(oct)2),在真空干燥和磁力搅拌将原料于70℃下预干燥1小时。快速升温到130-170℃,并在该温度下反应3~9h。产物经二氯甲烷和无水乙醚2-3次溶解沉淀提纯处理,真空干燥后得到聚乳酸样品,标记为pdlla,放置于-20℃冰箱中保存待用。得到的产物数均分子量(mn)为136.93kda,重均分子量(mw)为213.29kda,聚合物分散性指数(pdi)为1.56,转化率73.56%。
实施例3
立抅复合型丙交酯共聚合成聚乳酸
将比例为80:20的左旋丙交酯(lla)和外消旋丙交酯(dlla)装入施兰克瓶中,加入适量催化剂—辛酸亚锡(sn(oct)2),在真空干燥和磁力搅拌将原料于70℃下预干燥1小时。快速升温到130-170℃,并在该温度下反应3~9h。产物经二氯甲烷和无水乙醚2-3次溶解沉淀提纯处理,真空干燥后得到聚乳酸样品,标记为pdlla,放置于-20℃冰箱中保存待用。得到的产物数均分子量(mn)为118.96da,重均分子量(mw)为181.37kda,聚合物分散性指数(pdi)为1.52,转化率82.57%。
实施例4
将一定比例的左旋丙交酯(lla)和外消旋丙交酯(dlla)装入施兰克瓶中,加入适量催化剂—辛酸亚锡(sn(oct)2),在真空干燥和磁力搅拌将原料于70℃下预干燥1小时。快速升温到130-170℃,并在该温度下反应一定时间。产物经二氯甲烷和无水乙醚2-3次溶解沉淀提纯处理,真空干燥后得到聚乳酸样品,标记为pdlla,放置于-20℃冰箱中保存待用。
其中左旋丙交酯(lla)和外消旋丙交酯(dlla)的比例分别为:100:0、80:20,70:30,50:50,30:70,20:80,0:100,分别记为编号:pdl_100:0、pdl_80:20、pdl_70:30、pdl_50:50、pdl_30:70、pdl_20:80、pdl_0:100。
使用傅立叶变换红外光谱仪对不同单体比例的pdl样品进行了红外光谱测试。采用的是热熔成膜的制样方法。测试的波长范围为4000~500cm-1。结果如图1所示。从图1a可以看出,所制备的pdl聚合物在1750cm-1吸收峰为c=o的伸缩振动,在1270cm-1吸收峰为c-o-c的伸缩振。在1450cm-1吸收峰为–ch弯曲振动。2970cm-1、2872cm-1、3000cm-1的吸收峰对应着聚乳酸的c–h伸缩振动和-ch3伸缩振动。核磁共振氢谱(图1b)表明聚合物呈现出聚乳酸典型的特征化学位移(5.15为-ch上面氢的化学位移,1.56为-ch3的化学位移)。红外光谱图和核磁共振波谱数据表明pdl聚合物制备成功。
对样品进行傅立叶变换的核磁测试,测试共聚物溶解在氘代氯仿(cd3cl)中、四甲基硅烷(tms)为内标物,进行了核磁共振光谱测试。结果如图2所示。凝胶渗透色谱数据表明,单体比率的变化对所合成的聚合物的分子量影响比较明显。整体来说,所制备的聚合物的分子量基本能维持在100kda左右,其中当dlla/lla比率为30:70时数均分子量最大,可达270kda。影响分子量变化的因素很多,如体系的粘度、残存含水量、杂质等均会对分子量较大影响。单体比率对聚乳酸分子量影响的具体原因和机理有待进一步研究。
对编号为pdl_100:0、pdl_80:20、pdl_70:30、pdl_50:50、pdl_30:70、pdl_20:80、pdl_0:100样品进行gpc测试,测试其分子量及其分布。凝胶渗透色谱仪是waters1515,流动相采用的四氢呋喃(thf),流速为1ml/min。标准样为单分散的聚苯乙烯(ps)。结果如图2所示。
测试的样条先使用平板硫化机180℃热压成型,厚度为0.5mm的薄膜,之后使用裁刀制成。聚乳酸的拉伸的力学性能测试方法参考的是国标gb/t1040-2006,样条为哑铃型,标距为25mm,厚度为0.5mm,拉伸的速率是10mm/min,测试环境温度25℃,相对湿度50%,每组测试的样品个数为5个。结果如图3所示。plla和pdlla的拉伸强度分别为55mpa和45mpa,前者拉伸强度更大可能归因于其较高的规整度和较好的结晶性能。当dlla单体含量超过20%时,立构复合体聚乳酸的拉伸强度明显下降,这表明dlla的引入严重打乱了plla的规整度和结晶有序性。总体来说,dlla含量较大的聚合物的断裂伸长率会有所降低,原因可能是高分子链在晶区与非晶区相互穿插缠绕,晶区可以起到物理交联点的作用,分散应力,而无规线团的存在可允许分子链更灵活伸展。因此一定程度的结晶性有利于分子拉伸取向,提高材料的断裂伸长率。以上结果表明,不同旋光构象的单体组合可用于具有不同力学性能的立构复合体聚乳酸的可控构建。
- 还没有人留言评论。精彩留言会获得点赞!