一类化合物、其制备方法及其作为检测丙酮醛近红外二区荧光探针的应用与流程
本发明涉及一类化合物、其制备方法及其作为检测丙酮醛近红外二区荧光探针的应用。
背景技术:
丙酮醛(methylglyoxal,mgo)是一种化学性质活泼的α-酮醛类化合物,为体内葡萄糖、脂肪酸及氨基酸活性二羰基代谢产物。丙酮醛是一种强效糖化试剂,可对多种生物大分子,如蛋白质和dna等进行糖基化修饰,产生毒性终产物(advancedglycationendproduct,ages),从而导致蛋白质功能紊乱,激活膜受体及引发促炎信号。文献报道表明mgo与老年性疾病、糖尿病并发症和慢性炎症密切相关(drugmetab.druginteract.2008,23:125-150;endocrine.2013,43:472-484;diabetologia.2001,44:129-146;aminoacids.2012,42:1133-1142.)。在高血糖、肾脏病变、氧化应激及尿毒症等患者体内,丙酮醛水平显著升高(schmidt,cell.2006,124,258–260.)。因此,对丙酮醛进行活体检测,有望对相关疾病的诊断及疗效评估提供辅助信息。
丙酮醛的常规检测方法有电化学方法、滴定法及色谱法。但这些方法或应用范围窄,或灵敏度低,或需溶解细胞,操作冗繁而受到限制(anal.chim.acta.2012,751:66-70;anal.bioanal.chem.2012,403:2577-2581;anal.methods.2015,7:2386–2390.)。近红外二区(nir-ii)响应性荧光探针(1000–1700nm)因其光子散射低、光信号衰减少、组织自荧光现象和体内干扰物质光吸收降低等优势,可实现更深组织及高信噪比的活体成像,在分子诊断及治疗方面极具优势。特别是,近来开发的基于电子给体-电子受体-电子给体(d-a-d)的有机小分子近红外二区荧光探针,其良好的代谢性质及生物相容性,在临床转化中显示出极大潜力(chem.soc.rev.2018,doi:10.1039/c8cs00234g)。因此,开发基于d-a-d型丙酮醛响应型近红外二区荧光探针,实现丙酮醛活体检测,对相关疾病诊断及疗效评估具有重要意义。
技术实现要素:
本发明的一方面提供了通式i所示的化合物或其盐:
其中,x为s、o或se;
r1,r2各自独立地为h、c1-c8烷基、c1-c8烷氧基、卤素,或,r1和r2与其相邻的c一起形成5-10元杂环基;
如,
其中,r3和r32各自独立地为h、c1-c8烷基、式-(ch2)n1-(och2ch2)n2-or的聚乙二醇基;n1和n2分别为0~500的整数,r选自h、c1-c8烷基、羟基、氨基、羧基、磺酸基、卤素;所述的式-(ch2)n1-(och2ch2)n2-or的聚乙二醇基可进一步被c1-c8烷基、羟基、氨基、巯基、卤素中的一种或多种所取代;
r4和r5各自独立地为h、c1-c8烷基、c1-c8烷基硅基、被c1-c8烷基取代或未取代的c6-c12芳基、被羧基、c1-c8烷基硅基c1-c8烷氧基羰基(例如,三甲基硅基乙氧基羰基)、磺酸基c1-c8烷基胺酰基(例如,-conh(ch2)3so3h)取代或未取代的c1-c8烷基c6-c12芳基、式-(ch2)n1-(och2ch2)n2-or的聚乙二醇基,n1和n2分别为0~500的整数,r选自h、c1-c8烷基、羟基、氨基、羧基、磺酸基、卤素,所述的聚乙二醇基可一步被c1-c8烷基、羟基、氨基、巯基、卤素中的一种或多种所取代;或,r4和r5与其相邻的n一起形成5-10元杂环基;
r6和r7各自独立地为h、被羟基、氨基、巯基、卤素取代或未取代的c1-c8烷基、c1-c8烷氧基、c1-c8烷基酰氧基。
进一步优选地,x为o、s或se;
r1,r2为h,或r1和r2与其相邻的c一起形成二氧六环基(例如,1,4-二氧六环基);
在一优选实施方式中,
r3和r32各自独立地为h、c1-c4烷基、-(ch2)n1-(och2ch2)n2-or的聚乙二醇基,其中,n1和n2各自独立地为1~4的整数,r为c1-c4烷基;进一步优选地,r3和r32各自独立地为h、-(ch2)n1-(och2ch2)n2-or的聚乙二醇基,其中,n1和n2均为2,r为甲基;
r4、r5各自独立地为苯基、c1-c4烷基硅基c1-c4烷氧基羰基苯基(例如,三甲基硅基乙氧基羰基丙基苯基)、磺酸基c1-c4烷基胺酰基c1-c4烷基苯基(例如,-c6h4(ch2)2conh(ch2)3so3h)、羧基c1-c4烷基苯基(例如,羧乙基苯基)、-(ch2)n1-(och2ch2)n2-or的聚乙二醇基,其中,n1和n2各自独立地为1~4的整数,r为c1-c4烷基;进一步优选地,r4、r5各自独立地为苯基、4-(3-羰基-3-(2-(三甲基硅基)乙氧基)丙基)苯基、4-(2-羧乙基)苯基、-(ch2)n1-(och2ch2)n2-or的聚乙二醇基,其中,n1和n2均为2,r为甲基。
在一优选实施方式中,通式i所示的化合物最优选为如下化合物:
术语“卤素”是指氟、氯、溴或碘。
术语“c1-c8烷基”是指链上具有1至8个碳原子的直链或支链饱和烃基,非限制性地包括甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基等。
术语“c1-c8烷氧基”是指上述链上具有1至8个碳原子的直链或支链烷基的氧醚基。例如,甲氧基、乙氧基、正丙氧基、仲丁氧基、叔丁基、正己氧基等。
术语“c1-c8烷基硅基”为结构rarbrcsi-,其中,ra、rb和rc中至少有一个为c1-c8烷基,其余为氢的基团,例如,三甲基硅烷、三乙基硅烷。
术语“磺酸基”是指-so3h。
术语“c1-c8烷基酰氧基”是指具有酰氧基-oc(o)rd取代基的c1-c8烷基,其中,rd包括h、“c1-c8烷基”、“c2-c8烯基”、“c2-c8炔基”、“杂环烷基”、“芳基”、“杂芳基”、“c1-c8烷基芳基”、“c1-c8烷基杂芳基”;“c1-c8烷基酰氧基”非限制地包括2-(乙酰氧基)乙基。
术语“c1-c8烷基硅基c1-c8烷氧基羰基”是指被c1-c8烷基硅基取代的具有1-8个碳原子的烷基-o-c(=o)-基团,非限制性地包括三甲基硅基乙氧基羰基。
术语“烷基芳基氨基”是指具有一个或两个烷基取代基(相互独立地选择)的-n(芳基)2或-nh(芳基)的基团,例如甲基苯基氨基,甲基二苯基氨基,乙基苯基氨基,正丙基苯基氨基、正丙基二苯基氨基、异丙基苯基氨基、叔丁基苯基氨基;
术语“c1-c8烷基硅基c1-c8烷氧基羰基c1-c8烷基氨基”是指被c1-c8烷基硅基c1-c8烷氧基羰基取代的烷基芳基氨基,非限制性地包括二(三甲基硅基乙氧基羰基丙基苯基)氨基。
术语“5-10元杂环基”是指含有一个或多个饱和和/或部分饱和环,其包括5至10个环原子,其中一个或多个环原子选自氮、氧或硫的杂原子,其余环原子为碳;例如,环氧丙烷、四氢呋喃基、吡咯烷基、四氢吡喃基、哌啶基、哌嗪基、吗啉基、硫代吗啉基。
术语“c6-c14芳基”是指包含6-14个环原子但环原子中不含杂原子的单环或双环芳香族环基,例如,苯基、萘基。
术语“c1-c8烷基c6-c10芳基”是指被c1-c8烷基取代的c6-c10芳基,非限制性的包括,苄基、甲基萘基、丙基苯基。
术语“5-18元杂芳基”是指包含5-18个环原子且在环原子中含有1-4个杂原子作为环成员的单价芳香环基团。杂原子可以选自氮、氧或硫。杂芳基可以是具有5-7个环原子的单环杂芳基,或者具有7-12个环原子的双环杂芳基。所述双环杂芳基中只要一个环是杂芳环即可,另一个可以是芳香环或非芳香环的,含杂原子的或不含杂原子的。杂芳基的例子包括但不限于吡咯基、吡唑基、咪唑基、噁唑基、吡啶基、嘧啶基、呋喃基、噻吩基、噻唑基、噻二唑基、四唑基、三氮唑基、异噁唑基、吲哚基、甲基吡啶鎓基噻唑基、苯并[d]恶唑、苯并[d]咪唑等。优选地,所述5-12元杂芳基选自吡啶基,嘧啶基,噁唑基,噻吩基,吲哚基,1,3-二氧代异吲哚基,1-氧代异吲哚基,咪唑基,咔唑基、苯并咔唑基、吡唑基,异噁唑基,苯并咪唑基和呋喃基。
本发明的另一方面提供了制备通式i所示的方法,所述方法包括以下步骤:
其中,x、r1、r2和
1)通式化合物a、化合物b经pd催化偶联反应得到通式化合物c;
2)通式化合物c在还原剂、溶剂存在的条件下,60–120℃反应,得到通式i所示的化合物。
优选地,步骤2)中,所述的还原剂为铁单质,所述溶剂为乙酸;所述反应的反应时间为2-10h。
优选地,步骤1)中,所述偶联反应使用四(三苯基膦)钯pd(pph3)4作为pd催化剂,化合物a与化合物b、四(三苯基膦)钯pd(pph3)4的物质的量之比为1:(2-2.5):(0.06-0.12);
所述的偶联反应为将化合物a与化合物b、四(三苯基膦)钯溶解于甲苯和1m碳酸钾水溶液的混合溶剂中,在氮气下保护下加热反应。
在另一优选例中,步骤1)中,所述偶联反应在110℃下加热进行。
在另一优选例中,步骤1)中,所述偶联反应时间为10–20h。
本发明的另一方面是提供通式i所示化合物或其盐作为近红外二区荧光探针在丙酮醛检测中的用途。本发明所述的通式i所示化合物或其盐可直接作为丙酮醛响应性荧光探针。
本发明的另一方面提供了包含本发明通式i所示化合物的组合物,所述组合物包括至少一种本发明化合物,和任选的药学上可接受的赋形剂。
本发明的另一方面是提供包含本发明通式i所示化合物的组合物作为近红外二区荧光探针在丙酮醛检测中的用途。
本发明的另一方面是提供一种丙酮醛的测定方法,包括将通式i所示的化合物或其盐用作丙酮醛的响应性荧光探针。
本发明的丙酮醛响应性荧光探针的使用方法没有特别的限定,可以和以往公知的丙酮醛响应性荧光探针同样使用。通常选自用上述通式i所示的化合物或其盐的物质溶解在生理盐水或缓冲液等水性介质或者乙醇、丙酮、乙二醇、二甲亚砜、二甲基甲酰胺等水混合性有机溶剂和水性介质的混合物等中,该溶液与丙酮醛反应,再测定荧光光谱即可。
所述的丙酮醛选自在疾病组织或体液中的丙酮醛,例如,血液、淋巴结及肿瘤中的丙酮醛。
附图说明
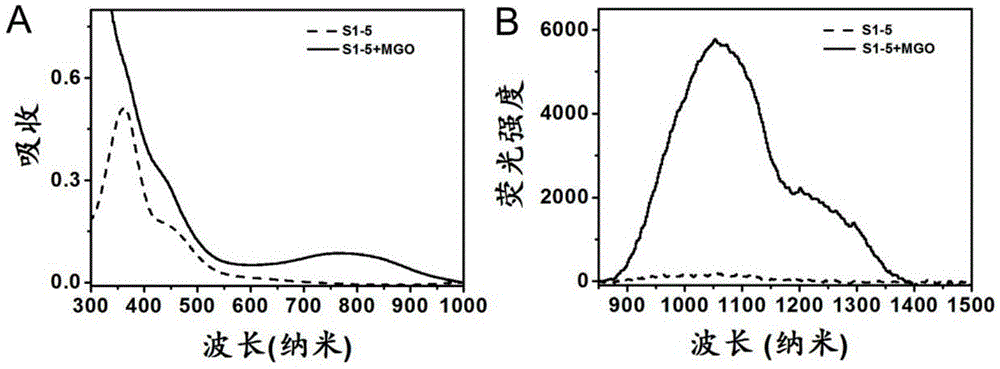
图1为化合物s1-5(20μm)与mgo反应前后的吸收光谱(a)和二区荧光光谱(b);测试条件为:10mm磷酸缓冲液pb(ph=7.4),50wt.%二甲基乙酰胺dmac,30μmmgo,37℃反应1h,激发波长808nm;
图2为化合物s1-6(20μm)与mgo反应前后的吸收光谱(a)和二区荧光光谱(b);测试条件为:10mmpb(ph=7.4),50wt.%dmac,30μmmgo,37℃反应1h,激发波长808nm;
图3为化合物s1-7(20μm)与mgo反应前后的吸收光谱(a)和二区荧光光谱(b);测试条件为:10mmpb(ph=7.4),50wt.%dmac,30μmmgo,37℃反应1h,激发波长808nm;
图4为化合物s1-8(20μm)与mgo反应前后的吸收光谱(a)和二区荧光光谱(b);测试条件为:10mmpb(ph=7.4),50wt.%dmac,30μmmgo,37℃反应1h,激发波长808nm;
图5为化合物是s1-7(100μm),尾静脉给药后5h成像图;测试条件:5wt.%dmac,10wt.%fbs,10mmpb(ph=7.4),给药剂量100μl,激发波长808nm。
具体实施方式
下面结合具体实施例对本发明做进一步阐述。这些实施例仅是出于解释说明的目的,而不限于不发明的范围和实质。
所有实施例中,1hnmr由avanceiii-300型核磁共振仪记录,化学位移以δ(ppm)表示;质谱由ms质谱-lcq-deca离子阱质谱仪(esi/lr)与ms质谱-q-tof四极杆飞行时间质谱仪(esi-hr)记录;反应检测中使用的薄层层析硅胶板(hsgf254)来自国药集团化学试剂有限公司;化合物分离选用国药集团化学试剂有限公司的200-300目硅胶。铁粉、1,4-二溴-2,3-二硝基苯并噻唑、三丁基(2,3-二氢噻吩并[3,4-b]-[1,4]二噁英-5-基)锡烷和双三苯基磷二氯化钯等试剂购买于国药集团化学试剂有限公司。
实施例1、化合物s1-1的合成
化合物c1制备参考文献方法(chem.sci.,2016,7:6203-6207)合成。然后将化合物c1(100mg,0.06mmol)溶入3ml醋酸,加入fe粉(100mg,1.79mmol),反应液升温至100℃,在氮气保护下,反应6h。待反应结束后,用饱和碳酸氢钠溶液调节反应液的ph值为7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体化合物s1-1,55mg,收率57%。1hnmr(300mhz,cdcl3)δ7.50(d,j=8.49hz,4h),7.32(m,4h),7.07(m,20h),4.19(t,j=8.43hz,8h),2.92(m,8h),2.61(m,8h),0.99(t,j=8.46hz,8h),0.05(s,36h).13cnmr(125mhz,cdcl3)δ174.56,152.20,149.05,147.38,147.05,140.71,136.96,135.09,131.15,130.66,129.12,128.12,126.16,124.35,123.82,108.59,64.13,37.45,31.84,31.04,18.89,0.00.esi-ms理论值为c82h100n6o8s3si4:1504.58,实测值为1504.45。
实施例2、化合物s1-2的合成
将化合物s1-1(10mg,0.0066mmol)溶于适量二氯甲烷中,加入三氟乙酸(二氯甲烷:三氟乙酸=10:1,v/v),室温搅拌5h。反应液减压浓缩,粗产物柱层析分离,得4mg黄色固体的化合物s1-2,收率54%。esi-ms理论值为c62h52n6o8s3:1104.3,实测值为1105.3[m+h]+。
实施例3、化合物s1-3的合成
化合物a2经两步合成:1,4-二溴-2,3-二硝基苯并噻唑(100.0mg,0.26mmol)、三丁基(2,3-二氢噻吩并[3,4-b]-[1,4]二噁英-5-基)锡烷(336.0mg,0.78mmol)和双三苯基磷二氯化钯(52.0mg,0.074mmol)加入到8.0ml重蒸甲苯中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得橙色固体的化合物i,126.6mg,收率96%。1hnmr(300mhz,cdcl3)δ6.77(s,2h),4.22(dd,j=11.5,5.5hz,8h).13cnmr(125mhz,cdcl3)δ152.63,143.07,142.52,141.21,120.28,105.49,104.61,64.53.
化合物i(80.0mg,0.158mmol)、nbs(62.0mg,0.347mmol)加入到3.0mldmf中,60℃下搅拌3.5h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得棕红色固体的化合物a2,103.0mg,收率98%。1hnmr(300mhz,dmso)δ4.27(d,j=27.0hz,8h).lr-ei-ms:[m+h]+m/z664.0。
化合物a2(108.0mg,0.151mmol)、化合物b1(50.2mg,0.075mmol)和四(三苯基膦)钯(9.0mg,0.0077mmol)加入到8.0ml重蒸甲苯和2.7ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体的化合物c3,76.9mg,收率61%。1hnmr(300mhz,cdcl3)δ7.62(d,j=8.8hz,4h),7.06(m,20h),4.29(d,j=24.9hz,8h),4.22–4.13(m,8h),2.91(t,j=7.8hz,8h),2.61(t,j=7.8hz,8h),1.04–0.92(m,8h),0.05(s,36h).13cnmr(125mhz,cdcl3)δ176.71,156.26,151.20,149.11,146.55,140.50,139.31,132.84,130.97,128.48,126.10,123.07,105.53,68.09,66.30,39.74,34.03,20.97,2.17。
化合物c3(50mg,0.03mmol)溶于2ml醋酸中,加入fe粉(33.5mg,0.60mmol),反应液升温至100℃,在氮气保护下反应6h。待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得23mg黄色固体的化合物s1-3,收率48%。1hnmr(300mhz,cdcl3)δ7.60(d,j=8.4hz,4h),7.05(m,20h),4.34(d,j=8.6hz,12h),4.24–4.03(m,8h),2.91(t,j=7.6hz,8h),2.60(t,j=7.8hz,8h),1.07–0.85(m,8h),0.05(s,36h).13cnmr(125mhz,cdcl3)δ176.54,154.53,150.05,149.14,143.20,140.46,138.68,132.54,130.51,128.13,122.56,109.59,17.84,68.16,66.07,39.55,33.81,20.75,1.96。
实施例4、化合物s1-4的合成
将化合物s1-3(10mg,0.0062mmol)溶于适量二氯甲烷中,加入三氟乙酸(二氯甲烷:三氟乙酸=10:1,v/v),室温搅拌5h。反应液减压浓缩,粗产物柱层析分离,得5mg黄色固体的化合物s1-4,收率66%。esi-ms理论值为c66h56n6o12s3:1220.3,实测值为1221.3[m+h]+。
实施例5、化合物s1-5的合成
化合物b2制备参考文献方法(sensorsandactuatorsb.2018,267:403–411.)化合物a1(100.0mg,0.183mmol)、化合物b2(161.7mg,0.366mmol)和四(三苯基膦)钯(25.41mg,0.022mmol)加入到6.0ml重蒸甲苯和3.0ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体的化合物c5,65.4mg,收率35%。1hnmr(300mhz,cdcl3)δ7.48(m,6h),7.33(t,j=7.6hz,4h),7.24(d,j=4.1hz,2h),7.19(d,j=7.7hz,4h),7.09(t,j=7.4hz,2h),6.93(d,j=8.5hz,4h),3.97(t,j=6.3hz,4h),3.71(t,j=6.0hz,4h),3.62(m,12h),3.58–3.48(m,4h),3.37(s,6h).esi-ms理论值为c52h52n6o10s3:1016.3,实测值为1017.0[m+h]+。
将化合物c5(56.6mg,0.056mmol)溶于2ml醋酸中,加入fe粉(62.2mg,1.11mmol),反应液加热至100℃,在氮气保护下反应6h;待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体的化合物s1-5,31.2mg,收率59%。1hnmr(300mhz,cdcl3)δ7.52(d,j=8.7hz,4h),7.33–7.29(m,8h),7.15(d,j=7.6hz,4h),7.06–6.97(m,6h),4.52(s,4h),3.97(t,j=6.4hz,4h),3.71(t,j=6.3hz,4h),3.67–3.58(m,12h),3.56–3.50(m,4h),3.37(s,6h).13cnmr(125mhz,cdcl3)δ150.81,147.65,147.28,146.21,139.29,133.28,129.69,129.46,126.84,126.18,123.01,122.75,122.01,119.15,107.16,71.95,70.76,70.68,70.61,68.19,59.07,51.63.esi-hrms理论值为c52h57n6o6s3:957.3496,实测值为957.3505[m+h]+。
实施例6、化合物s1-6的合成
化合物a2(100.0mg,0.151mmol)、化合物b2(133.4mg,0.302mmol)和四(三苯基膦)钯(20.95mg,0.018mmol)加入到3.0ml重蒸甲苯和1.0ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体的化合物c6,156.8mg,收率91%。1hnmr(300mhz,cdcl3)δ7.64(d,j=8.8hz,3h),7.31(t,j=7.9hz,4h),7.16(d,j=7.8hz,4h),7.08–7.03(m,3h),6.97(d,j=8.8hz,4h),4.37–4.17(m,8h),3.98(t,j=6.3hz,4h),3.71(t,j=6.5hz,4h),3.66–3.59(m,12h),3.57–3.50(m,4h),3.37(s,6h).esi-ms理论值为c56h56n6o14s3:1132.30,实测值为1133.1[m+h]+。
将化合物c6(74mg,0.065mmol)溶于2ml醋酸中,加入fe粉(73mg,1.31mmol),升温至100℃,在氮气保护反应6h。待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体的化合物s1-6,36.2mg,收率52%。1hnmr(300mhz,cdcl3)δ7.63(d,j=8.8hz,4h),7.31–7.25(m,4h),7.11(d,j=8.4hz,4h),7.01–6.96(m,6h),4.36(d,j=5.1hz,4h),4.32(d,j=4.6hz,4h),3.96(t,j=6.5hz,4h),3.70(t,j=6.4hz,4h),3.65–3.59(m,12h),3.56–3.51(m,4h),3.37(s,6h).13c-nmr(125mhz,cdcl3)δ151.17,147.50,146.64,140.13,139.48,136.84,129.34,127.30,125.26,122.16,122.11,119.76,119.35,105.83,104.43,71.96,70.75,70.67,70.60,68.21,64.92,64.60,59.05,51.57.esi-hrms理论值为c56h61n6o10s3:1073.3606,实测值为1073.3604[m+h]+。
实施例7、化合物s1-7的合成
化合物b3经两步合成。3-溴咔唑(250mg,1.016mmol)、氢氧化钾(105.0mg,1.86mmol)与适量18-冠-6溶于5mldmso中,室温搅拌1h后,加入对甲基三甘醇单甲醚苯磺酸酯(500mg,1.58mmol),室温搅拌过夜。向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得无色油状物的化合物ii,419.6mg,产率53%。1hnmr(300mhz,cdcl3)δ8.14(d,j=1.8hz,1h),7.97(d,j=7.8hz,1h),7.50–7.37(m,3h),7.28–7.19(m,2h),4.34(t,j=5.8hz,2h),3.77(t,j=5.8hz,2h),3.48–3.36(m,8h),3.34(s,3h)。
化合物ii(1.56g,3.98mmol)、频哪醇联硼酸酯(1.22g,4.79mmol)、乙酸钾(938mg,9.57mmol)与催化剂二(三苯基膦)二氯化钯(279.2mg,0.398mmol)溶于15mldmf中,氩气保护,至80℃油浴锅过夜反应。向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得浅黄色油状物的化合物b3,1.62g,产率93%。1hnmr(300mhz,cdcl3)δ8.59(s,1h),8.12(d,j=7.7hz,1h),7.92(d,j=8.3hz,1h),7.49–7.41(m,3h),7.27–7.22(m,1h),4.50(t,j=6.0hz,2h),3.86(t,j=6.0hz,2h),3.53–3.37(m,8h),3.33(s,3h),1.41(s,12h).esi-ms理论值为c25h34bno5:439.3,实测值为462.3[m+na]+。
化合物a1(100.0mg,0.183mmol)、化合物b3(160.9mg,0.366mmol)和四(三苯基膦)钯(25.41mg,0.022mmol)加入到3.0ml重蒸甲苯和1.0ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体的化合物c7,147.5mg,收率80%。1hnmr(300mhz,cdcl3)δ8.37(s,2h),8.13(d,j=7.7hz,2h),7.77(d,j=7.6hz,2h),7.55–7.42(m,10h),7.30–7.26(m,2h),4.51(t,j=5.8hz,4h),3.89(t,j=5.7hz,4h),3.54–3.47(m,12h),3.43–3.41(m,4h),3.33(s,6h).esi-ms理论值为c52h48n6o10s3:1012.3,实测值为1035.0[m+na]+。
将化合物c7(120mg,0.12mmol)溶于3ml醋酸中,加入fe粉(132.8mg,2.37mmol),升温至100℃,于氮气保护下反应6h;待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体的化合物s1-7,90mg,收率80%。1hnmr(300mhz,cdcl3)δ8.37(s,2h),8.13(d,j=7.7hz,2h),7.78(d,j=8.7hz,2h),7.49–7.45(dd,j=7.2,3.5hz,8h),7.38(d,j=3.6hz,2h),7.27–7.22(m,2h),4.57(s,4h),4.51(t,j=5.9hz,4h),3.89(t,j=5.8hz,4h),3.53–3.48(m,12h),3.45–3.42(m,4h),3.34(s,6h).13c-nmr(125mhz,cdcl3)δ150.88,147.36,141.09,140.41,139.37,133.48,129.72,126.07,125.61,124.26,123.40,122.91,122.29,120.51,119.38,117.82,109.40,109.15,107.20,71.87,71.02,70.64,70.55,69.37,59.00,43.35.esi-hrms理论值为c52h53n6o6s3:953.3183,实测值为953.3192[m+h]+。
实施例8、化合物s1-8的合成
化合物a2(100.0mg,0.151mmol)、化合物b3(132.75mg,0.302mmol)和四(三苯基膦)钯(20.95mg,0.018mmol)加入到3.0ml重蒸甲苯和1.0ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体的化合物c8,110mg,收率64.5%。1hnmr(300mhz,cdcl3)δ8.51(s,2h),8.15(d,j=7.8hz,2h),7.90(d,j=8.9hz,2h),7.51–7.47(m,6h),7.27(m,2h),4.51(t,j=5.4hz,4h),4.40(brs,4h),4.31(brs,4h),3.89(t,j=5.5hz,4h),3.50(m,12h),3.44(m,4h),3.34(s,6h).esi-ms理论值为c56h52n6o14s3:1128.3,实测值为1129.8[m+h]+。
将化合物c8(44mg,0.04mmol)溶于2ml醋酸中,加入fe粉(43.7mg,0.78mmol),升温至100℃,于氮气保护下反应6h;待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体的化合物s1-8,29.2mg,收率70%。1hnmr(300mhz,cdcl3)δ8.48(s,2h),8.14(d,j=7.9hz,2h),7.89(d,j=10.0hz,2h),7.50–7.43(m,6h),7.26–7.21(m,2h),4.51(t,j=5.8hz,4h),4.47–4.41(m,8h),4.39–4.34(m,4h),3.89(t,j=5.8hz,4h),3.57–3.48(m,12h),3.47–3.41(m,4h),3.34(s,6h).13c-nmr(125mhz,cdcl3)δ151.27,140.99,140.19,139.75,139.58,136.69,125.86,124.78,124.08,123.20,123.04,120.58,120.43,119.20,118.46,109.07,109.04,105.83,104.49,71.87,71.03,70.64,70.57,69.34,65.02,64.68,59.01,43.28.esi-hrms理论值为c56h57n6o10s3:1069.3293,实测值为1069.3294[m+h]+。
实施例9、化合物s1-9的合成
化合物s1-9制备参考文献(naturecommunication.2017,8:15269.)化合物s1-2(50mg,0.045mmol)、牛磺酸(219.6mg,1.76mmol)、n,n-二异丙基乙胺(227.5mg,1.76mmol)溶于700uldmso中,反应10min后,加入o-苯并三氮唑-四甲基脲六氟磷酸酯(220.1mg,0.58mmol),于氮气保护下过夜反应。次日加入等量水搅拌4h,淬灭反应。加入二氯甲烷,收集水层,于反相c18硅胶柱层析分离,得黄色固体35mg,收率50%。1hnmr(400mhz,dmso-d6)δ7.80(t,j=5.5hz,4h),7.60(d,j=8.6hz,4h),7.47(d,j=3.6hz,2h),7.32(d,j=3.6hz,2h),7.17(d,j=8.4hz,8h),6.96(d,j=8.6hz,8h),3.32(t,j=7.7hz8h),2.78(t,j=7.7hz,8h),2.54(d,j=7.7hz,8h),2.35(t,j=7.8hz,8h).13cnmr(126mhz,dmso-d6)δ171.46,150.46,147.44,145.29,144.01,140.56,136.83,134.95,129.92,127.65,126.88,124.87,123.04,122.59,102.16,51.12,37.58,35.97,30.99.
实施例10、化合物s1-10的合成
化合物b4经三步合成。将六甘醇单甲醚(1g,3.37mmol)溶于5ml二氯甲烷中,加入三乙胺(409.5mg,4.05mmol)、对甲基苯磺酰氯(769.3mg,4.05mmol),并于60℃油浴锅反应过夜。次日反应液浓缩,粗产物柱层析分离,得无色油状物iii966mg,产率90%。1hnmr(400mhz,cdcl3)δ7.77(d,j=7.6hz,2h),7.32(d,j=7.4hz,2h),4.13(s,2h),3.65–3.52(m22h),3.35(s,3h),2.42(s,3h).
3-溴咔唑(595mg,2.42mmol)、氢氧化钾(247.8mg,4.43mmol)与适量18-冠-6溶于5mldmso中,室温搅拌1h后,加入iii(1.2g,3.77mmol),室温搅拌过夜。向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得无色油状物iv1.14g,产率90%。1hnmr(400mhz,cdcl3)δ8.17(s,1h),8.01(d,j=7.7hz,1h),7.53–7.22(m,5h),4.44(m,2h),3.83(m,2h),3.65–3.38(m,20h),3.38(s,3h).
化合物iv(1.56g,3.98mmol)、频哪醇联硼酸酯(1.22g,4.79mmol)、乙酸钾(938mg,9.57mmol)与催化剂二(三苯基膦)二氯化钯(279.2mg,0.398mmol)溶于15mldmf中,氩气保护,至80℃油浴锅过夜反应。向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得浅黄色油状物b41.62g,产率93%。1hnmr(400mhz,cdcl3)δ8.58(s,1h),8.16(s,1h),7.99(d,j=8.9hz,1h),7.67(d,j=8.1hz,1h),7.48–7.43(m,3h),7.22(m,1h),4.43(m,2h),3.83(m,2h),3.66–3.55(m,10h),3.54–3.44(m,10h),3.35(s,3h),1.21(s,12h).
化合物a1(200.0mg,0.366mmol)、化合物b4(418.73mg,0.732mmol)和四(三苯基膦)钯(50.8mg,0.044mmol)加入到6.0ml重蒸甲苯和2.0ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体c1084.1mg,收率36%。1hnmr(400mhz,cdcl3)δ8.41(s,2h),8.17(d,j=7.7hz,2h),7.82(d,j=8.5hz,2h),7.59–7.47(m10h),7.33–7.29(m,2h),4.55(t,j=5.9hz,4h),3.92(t,j=5.9hz,4h),3.66–3.50(m,40h),3.39(s,6h).
将化合物c10(84.1mg,0.066mmol)溶于3ml醋酸中,加入fe粉(73.66mg,1.32mmol),升温至90℃,于氮气保护下反应6h;待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体30mg,收率37.5%。1hnmr(400mhz,cdcl3)δ8.38(s,2h),8.18–8.08(m,2h),7.86–7.74(m,2h),7.61–7.35(m,10h),7.29-7.27(m,2h),4.65(s,4h),4.51(m,4h),3.88(m,4h),3.74–3.43(m,40h),3.39(s,6h).13cnmr(126mhz,cdcl3)δ150.87,147.30,141.07,140.43,139.44,133.55,129.73,126.07,125.63,124.25,123.39,122.91,122.29,120.50,119.37,117.79,109.45,109.14,106.96,71.92,71.04,70.59,70.53,70.48,69.38,59.01,43.37.
实施例11、化合物s1-11的合成
化合物b5经三步合成。冰浴下,将对甲基苯磺酰氯(750mg,3.95mmol)与十二甘醇单甲醚(2g,3.57mmol)溶于四氢呋喃中,加入氢氧化钠(357mg,8.93mmol)的50%水溶液,加毕,转移至室温反应过夜。次日向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得无色油状物v1.86g,产率73%。1hnmr(400mhz,cdcl3)δ7.70(d,j=5.7hz,2h),7.27(d,j=5.7hz,2h),4.07(m,2h),3.56(m,46h),3.28(s,3h),2.36(s,3h).
3-溴咔唑(620mg,2.52mmol)与化合物v(1.8g,2.52mmol)溶于适量四氢呋喃,加入氢氧化钠(100mg,2.52mmol)的50%氢氧化钠溶液。置于油浴锅中回流反应过夜。次日向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得无色油状物vi1.7g,产率85.8%。1hnmr(400mhz,cdcl3)δ8.15(s,1h),8.00(d,j=7.8hz,1h),7.54–7.40(m,3h),7.34(m,1h),7.22(m,1h),4.44(t,j=5.4hz,2h),3.82(t,j=5.6hz,2h),3.63–3.48(m,44h),3.36(s,3h).
化合物vi(1.7g,2.15mmol)、频哪醇联硼酸酯(656.4mg,2.58mmol)、乙酸钾(506mg,5.16mmol)与催化剂二(三苯基膦)二氯化钯(150.64mg,0.215mmol)溶于15mldmf中,氩气保护,至80℃油浴锅过夜反应。向反应液中加入水,乙酸乙酯萃取。有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得浅黄色油状物b51.5g,产率83%。1hnmr(400mhz,cdcl3)δ8.58(s,1h),8.11(d,j=7.1hz,1h),7.91(d,j=8.2hz,1h),7.49–7.43(m,3h),7.26–7.21(m,1h),4.50(t,j=5.9hz,2h),3.86(t,j=5.9hz,2h),3.66–3.48(m,44h),3.37(s,3h),1.37(s,12h).
化合物a1(100.0mg,0.183mmol)、化合物b5(306,2mg,0.366mmol)和四(三苯基膦)钯(25.41mg,0.022mmol)加入到3.0ml重蒸甲苯和1.0ml1m碳酸钾水溶液中,氮气保护,回流反应12h。tlc检测反应完毕后加水及乙酸乙酯萃取,有机层依次用水、饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得蓝色固体c11150mg,收率45.5%。1hnmr(400mhz,cdcl3)δ8.40(s,2h),8.17(d,j=8.7hz,2h),7.81(d,j=8.7hz,2h),7.61–7.42(m,10h),7.31(m,2h),4.55(t,j=5.9hz,4h),3.92(t,j=5.9hz,4h),3.69–3.51(m,88h),3.39(s,6h).
将化合物c11(100mg,0.055mmol)溶于5ml醋酸中,加入fe粉(62.1mg,1.11mmol),升温至100℃,于氮气保护下反应6h;待反应结束后,用饱和碳酸氢钠溶液调节反应液ph至7~8,二氯甲烷萃取三次,合并有机相。饱和食盐水洗涤,无水硫酸钠干燥。抽滤,滤液减压浓缩,粗产物柱层析分离,得黄色固体35mg,收率36%。1hnmr(400mhz,cdcl3)δ8.40(s,2h),8.15(d,j=7.7hz,2h),7.81(d,j=7.8hz,2h),7.56–7.40(m,10h),7.30-7.26(m,2h),4.54(t,j=5.8hz,4h),3.91(t,j=5.8hz,4h),3.71–3.48(m,88h),3.38(s,6h).13cnmr(126mhz,cdcl3)δ150.85,147.18,141.06,140.43,139.57,133.70,129.69,126.06,125.66,124.24,123.38,122.91,122.28,120.50,119.37,117.77,109.46,109.14,106.50,71.92,71.05,70.54,69.40,59.01,43.38.esi-hrms理论值:c88h124n6o24s3na:1767.7721,实测值1767.7745[m+na]+.
实施例12、探针检测mgo的紫外和二区荧光光谱检测方法:
20μm探针(如s1-5、s1-6、s1-7和s1-8)溶于50%二甲基乙酰胺dmac中,在10mm磷酸缓冲液pb(ph=7.4)的缓冲中加入30μmmgo37度反应1h,用1cm常量比色皿,体积2ml,分别测吸收光谱与二区荧光光谱。吸收检测波长范围:300-1000nm;荧光光谱参数:808激光器,功率:2w,激发狭缝宽度:5nm,发射狭缝宽度:10nm,收集波长:850-1500nm。仪器名称:安捷伦cary60紫外-可见分光光度计,爱丁堡fls980荧光光谱仪。结果如图1-4所示,探针s1-5、s1-6、s1-7和s1-8在pb缓冲液中,对mgo具有很好的响应,分别在650nm至900nm之间产生一个新的最大吸收峰,于1000nm至1100nm之间产生一个新的最大二区荧光发射峰。
实施例13、探针检测mgo的乳腺癌mcf-7移植瘤小鼠活体成像方法
肿瘤瘤内注射乙二醛酶1(glo1)抑制剂,过夜12h,以便在瘤内诱导产生mgo;然后,将本发明制备的化合物s1-7(探针s1-7)溶于5wt.%二甲基乙酰胺(dmac)中,10mm磷酸缓冲液pb(ph=7.4)调节探针浓度至100μm,尾静脉注射100μl,5h后荧光成像。荧光光谱参数:808激光器,功率:2w,激发狭缝宽度:5nm,发射狭缝宽度:10nm,收集波长:1000-1250nm。结果如图5所示,探针s1-7与肿瘤组织中高表达的mgo反应,产生二区荧光发射峰,乳腺癌组织呈高二区荧光信号。
- 还没有人留言评论。精彩留言会获得点赞!