一种基于NHPI官能化的多羧酸有机配体及合成方法与流程
本发明涉及一种基于nhpi官能化的多羧酸有机配体,本发明进一步涉及该多羧酸配体的合成方法,属于有机化工合成技术领域。
背景技术:
近年来,n-羟基邻苯二甲酰亚胺(nhpi)及其类似物在有机氧化反应中的应用引起了人们极大的关注。它们作为电化学氧化基质或催化剂广泛应用于芳烃、烷烃、烯烃、炔烃、醇类、醚、酰胺和缩醛类的氧化反应中。特别是辅以过渡金属盐时,能在常压和25~100℃的温和条件下实现许多有机化合物的催化氧化,是比较理想的绿色氧化剂。但作为均相的小分子催化剂,n-羟基邻苯二甲酰亚胺(nhpi)与其他小分子催化剂一样存在着无法回收利用,增大反应后处理难度等问题。
为了解决这类问题,相关研究人员通过化学合成的方法将nhpi固载在一些特制的交联共聚微球表面,从而赋予了nhpi非均相的特性。例如“一种简捷、高效的制备固相催化剂微球cps-nhpi的方法”(中国发明专利,申请号:201510675234.0)采用friedel-crafts酰基化反应和酰亚胺反应两步大分子反应,在微球cps表面实现了催化基团nhpi的同步合成与固载,得到催化剂微球cps-nhpi。这种方法虽然一定程度上实现了nhpi的重复利用性,但由于其较低的固载量,以及只能在表面实现固载从而使其难以实现理想的催化效果。“一种聚合物微球固载n-羟基邻苯二甲酰亚胺催化剂的制备方法”(中国发明专利,申请号:201410206270.8)通过新制作中间产物醛基化改性微球gma/mma-al;键合有邻苯二甲酸基团的改性微球gma/mma-pa;固载有邻苯二甲酸酐ppa的微球gma/mma-ppa;最终得到固载有n-羟基邻苯二甲酰亚胺基团的功能微球,并用其进行催化氧化性能的验证。这类固载化的交联微球虽然也实现了nhpi的重复利用性,但同样由于其较低的固载量,以及只能在表面实现固载,从而使其难以实现理想的催化效果。
鉴于金属有机框架材料(mofs)固有的多孔性以及优异的非均相催化性能,相关科研人员通过将小分子的nhpi限域在金属有机框架材料(mofs)的孔道内,实现其非均相催化的性能研究。例如hermenegildo等人利用fe3+和均苯三甲酸形成的金属有机框架材料对nhpi进行限域作用,使其固定在该mofs材料的孔道内,从而达到非均相催化的目的(aerobicoxidationofbenzylaminestobenzyliminescatalyzedbymetal-organicframeworksolids,chemcatchem,2010,1438-1443.)。然而这种方法无法将nhpi完全限域在其孔道内,在催化过程中nhpi会不断的泄漏,导致其重复利用性很差,也很难达到目标的催化效果。
技术实现要素:
本发明的目的在于,提供一种可被用来制备一系列三维多孔且孔道内裸露着大量以共价键连接,而且可产生氮氧自由基位点的n-羟基酰亚胺官能团的功能性材料的多羧酸有机配体。用该配体制备的金属有机框架材料(mofs)催化效果好,能多次重复利用。本发明另外一个目的是公开该配体的合成方法。
为了达到上述目的,本发明从有机配体的功能化设计出发,通过有机合成的方法将n-羟基邻苯二甲酰亚胺基团直接引入用来构筑多孔骨架材料的多羧酸有机配体中,这样使得用该类配体制备的金属有机框架材料(mofs)不仅在孔道内富含活性催化位点,而且以共价键连接的功能基团也可以长时间稳定存在,这样就同时解决了固载量低和回收利用性的问题,而目前为止还没有任何相关的报道。
本发明致力于合成一种能够用来制备三维多孔且在孔道中能够直接以共价键的方式裸露出具有催化活性的n-羟基邻苯二甲酰亚胺官能团的多羧酸有机配体。所述n-羟基邻苯二甲酰亚胺修饰的多羧酸有机配体的结构式大致如下所示:
式中,\cooh为单-cooh或双-cooh;
其中:单-cooh是在苯环上的4位上;双-cooh是在苯环上的两个间位上;具体结构如下:
上述(a)为4,4'-(2-羟基-1,3-二氧代异吲哚啉-4,7-二基)二苯甲酸
上述(b)为5,5'-(2-羟基-1,3-二氧代异吲哚啉-4,7-二基)二异酞酸。
本发明的基于nhpi官能化的多羧酸有机配体的制备方法是:先按照摩尔比量取1,4-二溴-2,3-二甲基苯:甲基苯硼酸:钯催化剂:无机碱:去离子水=2:1:0.05:4:0.05,然后再将他们加入有机溶剂中,在n2保护下加热,温度为90-120℃,反应时间为8-12h,反应结束后过柱纯化;再按多甲基化合物上单个甲基:高锰酸钾=1:6的摩尔比称取两者加入到吡啶和去离子水的等比例混合溶液中,加热90-110℃,反应时间为15-20h,从而氧化甲基;反应结束后过滤、旋去吡啶、加稀盐酸酸化,干燥,即得多羧酸的产物,最后再将干燥后的多羧酸产物与盐酸羟胺置于有机溶剂吡啶溶液中加热反应,多羧酸产物:盐酸羟胺=1:25摩尔比,反应温度为90℃,反应时间为3h;反应结束后旋去溶剂,加入稀盐酸酸化,离心、水洗、干燥后即得最终n-羟基邻苯二甲酰亚胺(nhpi)修饰的多羧酸有机配体;
上述甲基苯硼酸为苯环上4位或3,5位带有甲基的苯硼酸;
上述钯催化剂为四三苯基磷钯;
上述碱是碳酸钠或碳酸钾;
上述有机溶剂为甲苯或1,4-二氧六环;
上述柱层析所用洗脱剂为石油醚;
上述稀盐酸酸化所用的稀盐酸为1mol/l稀盐酸溶液;
上述最后一步反应的具体后处理为先将吡啶溶液旋干,加入1mol/l稀盐酸溶液,超声片刻,离心,水洗,干燥即得n-羟基邻苯二甲酰亚胺(nhpi)官能化的多羧酸有机配体。
本发明具有如下的优点和有益效果:
1.本发明鉴于n-羟基邻苯二甲酰亚胺(nhpi)良好的分子氧氧化性能和应用,首次尝试将具有自由基活性的n-羟基邻苯二甲酰亚胺引入能够用来合成三维多孔材料的多羧酸有机配体中,从而实现有机配体的直接官能化;实验结果表明用该类配体制备的金属有机框架材料(mofs)具备良好的分子氧氧化性能和优异的重复利用性。
2.本发明通过大量工艺实验路线的设计探究,找出了简单合理又普遍适用的合成路线,不仅每一步都有着高达85%-96%的产率,而且通过不同数量羧基的引入,也使得以后用来形成三维多孔材料的种类得到丰富。
3.本发明方法最后利用生成的具有邻苯二甲酸的结构单元和盐酸羟胺直接反应得到了n-羟基邻苯二甲酰亚胺官能团,与传统工艺比较,省去了一般该类反应所需的先将二羧酸脱水成酸酐再与盐酸羟胺反应的步骤,过程上得到了简化,而且产物的纯度也得到了很大的提高,可以达到96%,这对于因为溶解性问题而较难纯化的多羧酸物质至关重要。
4.本发明从有机配体的结构设计出发,直接在多羧酸配体中引入了n-羟基邻苯二甲酰亚胺的官能团,而合成的配体可以用来制备一系列金属有机框架材料(mofs),而这类mofs材料因其高达90%-99%的优异的催化效率和回收再利用性等综合性能在催化领域有着很广泛的应用前景,因此,本发明在实现nhpi非均相催化的实验设计中开创了一种新的思路和可能。
附图说明
图1为本发明化合物a的核磁氢谱
图2为本发明化合物a的核磁碳谱
图3为本发明化合物b的核磁氢谱
图4为本发明化合物b的核磁碳谱
具体实施方式
下面结合附图和具体实施对本发明进行进一步说明。
实施例1
基于n-羟基邻苯二甲酰亚胺(nhpi)修饰的多羧酸有机配体(a):4,4'-(2-羟基-1,3-二氧代异吲哚啉-4,7-二基)二苯甲酸(简称h2ln-oh)的具体结构如下:
h2ln-oh的合成方法的反应式如下:
基于nhpi官能化的多羧酸有机配体(a)的制备:在n2保护下向100ml的三口烧瓶中加入6.3mmol1,4-二溴-2,3-二甲基苯、14.1mmol4-甲基苯硼酸、25.6mmol碳酸钠、0.2mmol四三苯基磷钯、30ml甲苯、6ml去离子水,再用氮气置换三次,然后在磁力搅拌下用油浴加热至90℃,反应6h。反应结束后,水洗处理,萃取,干燥后减压除去有机溶剂,产物经柱色谱(石油醚)的方法分离得到,产率为85%。再将1.75mmol所得产物、42mmol高锰酸钾、10ml吡啶、12ml蒸馏水加入100ml三口烧瓶中,然后在磁力搅拌下用油浴加热至100℃,反应回流15h,反应结束后,减压旋去有机溶剂,然后加水,超声片刻后通过硅藻土过滤掉mno2,加盐酸调ph至1即可析出大量白色粉末,离心收集并干燥,得到多羧酸产物,产率为97%。最后将1.98mmol四羧酸产物、4mmol盐酸羟胺、10ml吡啶加入50ml单口烧瓶中,在磁力搅拌下用油浴加热至90℃,反应6h。反应结束后减压旋去有机溶剂,然后加稀盐酸超声片刻,离心收集并干燥即可得到米白色的产品:基于nhpi官能化的多羧酸有机配体(a),产率为96%。
该产物多羧酸有机配体(a)的表征数据及谱图如下:
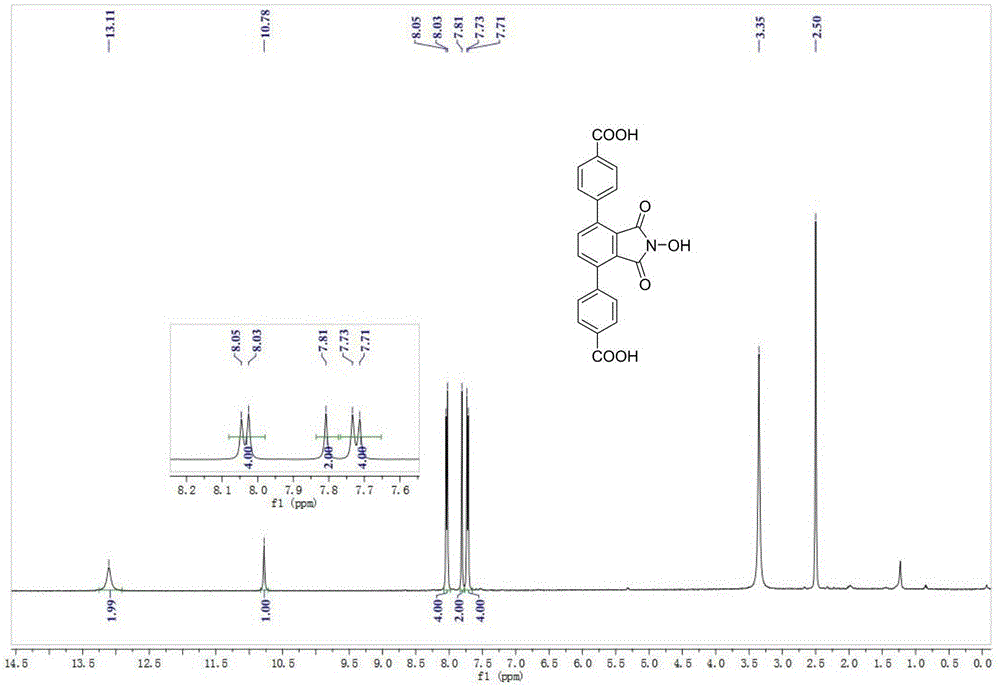
1hnmr(dmso-d6,400mhz):δ13.11(s,2h),10.78(s,1h),8.04(d,j=8.2hz,4h),7.81(s,2h),7.72(d,j=8.1hz,4h).13cnmr(151mhz,dmso)δ167.16,163.01,140.28,137.97,135.91,130.72,129.80,128.90,125.84.
请看图1,因为多羧酸有机配体(a)是对称的结构,分析结构可知会有5种不同的氢信号,所测核磁氢谱中7.22ppm处的双重峰对应着羧酸间位上的氢信号,7.81ppm处的单峰对应着中间苯环上的两个氢信号,8.04ppm处的双重峰对应着羧酸邻位上的氢信号,10.78ppm处的单峰对应着氮羟基上氢的特征信号峰,13.11ppm处的单峰对应着羧酸的活泼氢的特征信号峰,相应的氢原子的个数比和结构(a)完全一致(其中2.5ppm和3.3ppm处分别为所用氘代dmso的溶剂峰和水峰);
请看图2,因为(a)是对称的结构,分析结构可知会有9种不同的碳信号,所测核磁碳谱中167.16ppm处的信号峰对应着羧酸上的碳信号,163.01ppm处的信号峰对应着连接着亚胺的酮羰基上的碳信号,125.84-140.28ppm范围内的7个信号峰分别对应于三个苯环上7个不同化学环境的碳信号(其中39.50ppm处的七重峰为所用氘代dmso的碳信号的耦合裂分);
实施例2
n-羟基邻苯二甲酰亚胺(nhpi)修饰的多羧酸有机配体(b):5,5'-(2-羟基-1,3-二氧代异吲哚啉-4,7-二基)二异酞酸(简称h4ln-oh)的具体结构如下:
h4ln-oh的合成方法的反应式如下:
基于nhpi官能化的多羧酸有机配体(b)的制备:在n2保护下向100ml的三口烧瓶中加入6.3mmol1,4-二溴-2,3-二甲基苯、13.5mmol3,5-二甲基苯硼酸、25.6mmol碳酸钾、0.2mmol四三苯基磷钯、30ml1,4-二氧六环、6ml去离子水,再用氮气置换三次,然后在磁力搅拌下用油浴加热至90℃,反应6h。反应结束后,水洗处理,萃取,干燥后减压除去有机溶剂,产物经柱色谱(石油醚)的方法分离得到,产率为88%。再将1.9mmol所得产物、68mmol高锰酸钾、10ml吡啶、12ml蒸馏水加入100ml三口烧瓶中,然后在磁力搅拌下用油浴加热至100℃,反应回流15h,反应结束后,减压旋去有机溶剂,然后加水,超声片刻后通过硅藻土过滤掉mno2,加盐酸调ph至1即可析出大量白色粉末,离心收集并干燥,得到多羧酸产物,产率为94%。最后将1.8mmol六羧酸产物、4mmol盐酸羟胺、10ml吡啶加入50ml单口烧瓶中,在磁力搅拌下用油浴加热至90℃,反应6h。反应结束后减压旋去有机溶剂,然后加稀盐酸超声片刻,离心收集并干燥即可得到米白色的产品b,产率为97%。
该产物的表征数据如下:
1hnmr(dmso-d6,400mhz):δ13.44(s,4h),10.84(s,1h),8.57(s,2h),8.38(s,4h),7.89(s,2h).13cnmr(dmso-d6151mhz,)δ166.45,163.19,137.03,136.73,135.99,134.33,131.19,129.90,126.08.
请看图3,因为(b)是对称的结构,分析结构可知会有5种不同的氢信号,所测核磁氢谱中7.89ppm处的单峰对应着中间苯环上的两个氢信号,8.38ppm处的单峰对应着两端苯环上羧酸邻位的四个氢信号,8.57ppm处的单峰对应着两端两个羧基中间的氢的信号,10.84ppm处的单峰对应着氮羟基上氢的特征信号峰,13.44ppm处的单峰对应着羧酸的活泼氢的特征信号峰,相应的氢原子的个数比和结构(b)完全一致(其中2.5ppm和3.3ppm处分别为所用氘代dmso的溶剂峰和水峰);
请看图4,因为(b)是对称的结构,分析结构可知会有9种不同的碳信号,所测核磁碳谱中166.45ppm处的信号峰对应着羧酸上的碳信号,163.19ppm处的信号峰对应着连接着亚胺的酮羰基上的碳信号,126.08-137.03ppm范围内的7个信号峰分别对应于三个苯环上7个不同化学环境的碳信号(其中39.50ppm处的七重峰为所用氘代dmso的碳信号的耦合裂分);
实施例3
按1:1.2:25的摩尔比称取配体(a)、zrcl4、苯甲酸于玻璃瓶中,然后加入n,n’-二甲基甲酰胺(dmf)溶液,超声15分钟使溶液澄清,再将此溶液置于85℃的恒温烘箱中反应3天得到白色粉末沉淀,所得白色粉末沉淀用dmf洗涤后再用丙酮浸泡2天,100℃真空活化8小时,基于配体计算得到的晶体产率约为65%。pxrd测试结果表明所得白色粉末具有很好的结晶性,sem扫描结果显示,所得白色粉末为尺寸均一的正八面体结构,bet测试结果显示所得材料比表面积为2557.8386m2.g-1,从而证明了目标金属有机框架材料mofs(uio-68-nhpi)制备成功。再用所得mofs(uio-68-nhpi)作为非均相催化剂,o2作为氧源对伯醇和仲醇进行氧化,可得到相应的醛和酮,经过检测氧化产率一般可达到90%以上,实验结果表明所得uio-68-nhpi重复利用5次催化效果仍得到很好的保持。
实施例4
按1:2.5的摩尔比称取配体(b)和cu(no3)2'2.5h2o于玻璃瓶中,然后按2:1的比例加入n,n’-二甲基甲酰胺(dmf)和甲醇溶液,最后加入一滴hbf4,将混合溶液超声15分钟使配体和金属盐完全溶解,再将此澄清溶液置于85℃的恒温烘箱中反应18小时得到蓝色块状晶体,所得蓝色晶体用dmf洗涤后再用丙酮浸泡3天,100℃真空活化10小时,基于配体计算得到的晶体产率约为76%。pxrd测试结果表明所得蓝色晶体具有良好的结晶性,sem扫描结果显示所得晶体为尺寸均一的八面体结构,bet测试结果显示所得材料比表面积为2294.9205m2.g-1,从而证明了目标金属有机框架材料mofs(nott-nhpi)制备成功。再用所得mofs(nott-nhpi)作为非均相催化剂,o2作为氧源对伯醇和仲醇进行氧化,可得到相应的醛和酮,部分伯醇可得到相应的羧酸产物,检测结果表明氧化产率一般可达到95%以上,实验结果表明所得nott-nhpi重复利用7次催化效果仍得到很好的保持。
- 还没有人留言评论。精彩留言会获得点赞!