一种依托度酸光降解杂质及其制备方法与流程
本发明属于生物医药领域,具体涉及一种依托度酸光降解杂质及其制备方法。
背景技术:
依托度酸(etodolac)是吡喃糖羧酸类甾体抗炎药,通过阻断环氧合酶的活性,从而抑制了前列腺素(pg)的合成,具有抗炎、解热和阵痛作用。临床上用于治疗术后疼痛、缓解类风湿性关节炎和骨关节炎的症状。同时,药物广泛使用已经成为新兴的环境污染因素。对药品生产中各杂质项的研究和控制是非常必要的。
徐梅桔等(高效液相色谱法测定依托度酸的含量,广东化工,2013,40,(5)19-23)公开了将依托度酸分别进行酸破坏、碱破坏、氧化破坏、光破坏及热破坏试验,并对破坏试验后的成分进行检测,得到破坏试验后的高效液相色谱图,但是没有给出各相应杂质项。
杨婉花,(高效液相色谱法测定依法度酸缓释片中的有关物质,中国药房,2005年第16卷第17期,1330-1332)公开了几种依托度酸缓释片破坏试验的四种杂质及其高效液相色谱检测条件。
由以上可知,依托度酸的稳定性较差,受酸、碱、光和热等破坏,可产生几十种杂质,这些杂质的产生对临床用药存在潜在威胁,为了更好地实现该药品的经济和社会效益,标准中必须严控各项杂质,明确各杂质项是急需解决的问题。
技术实现要素:
本发明的目的在于提供一种依托度酸光降解杂质化合物,该化合物结构式如式ι所示。
本发明还提供了式ι杂质化合物的制备方法,依托度酸样品溶解后经光照破坏再纯化即得式ι依托度酸光降解杂质化合物。反应式如下所示。发明人进一步研究发现,式ι依托度酸光降解杂质化合物可能会发生进一步的降解,需要将光照破坏后的依托度酸溶液,进行纯化得到高纯度的式ι杂质化合物。然而采用正相色谱或反相色谱制备纯化式ι杂质化合物在后处理过程中易发生降解。发明人采用超临界流体色谱对式ι杂质化合物进行纯化,并改进流动相,能够有效提高式ι杂质化合物的收率和纯度,并进一步有效防止了式ι杂质化合物的再降解。
本发明所提供的式ι依托度酸光降解杂质化合物的制备方法包括如下步骤:
(1)、将依托度酸溶解在有机溶剂中,对依托度酸溶液进行光照破坏得依托度酸光照破坏液;
(2)、超临界流体色谱纯化步骤(1)所得依托度酸光照破坏液,收集含目标产物的洗脱流出液;
(3)、快速浓缩蒸干步骤(2)所得洗脱流出液即得式ι依托度酸光降解杂质化合物。
优选地,步骤(1)中所述有机溶剂为极性非质子溶剂或c1-c4烷醇的一种或几种,更加优选地,所述有机溶剂为四氢呋喃、乙酸乙酯、乙腈、乙醇或甲醇。
优选地,步骤(1)中所述的光照强度为3000~6000lx,光照时间为10~20h。
优选地,步骤(1)中所述的光照破坏是在有氧条件下进行的。更加优选地,所述的有氧条件是指先将依托度酸溶解在有机溶剂中,加入双氧水溶液再进行光照破坏;或将依托度酸溶解在有机溶剂中,光照破坏时通入空气或氧气。
优选地,步骤(1)中依托度酸样品溶解后,加入双氧水,所述双氧水的质量浓度为1~5%,添加体积为依托度酸溶液体积的0.5~1.5%。在依托度酸样品溶解后加入适量的双氧水溶液可增大依托度酸的光降解程度,使得依托度酸更大限度的转化为式i光降解杂质化合物。当双氧水的量过大时,依托度酸会更多的发生氧化破坏,抑制光降解反应发生。
优选地,步骤(2)中,所述超临界流体色谱填料为裸硅胶、二醇基、氨基型、酰胺型或两性离子型色谱填料;更加优选地超临界流体色谱填料为裸硅胶、二醇基硅胶基质填料。
优选地,超临界流体色谱控制范围为30~35℃,色谱系统控压范围为100~300bar。
所述步骤(2)中色谱流动相采用超临界co2、改性剂和添加剂。改性剂为极性非质子溶剂或c1~c4烷醇的一种或几种,添加剂为c1~c3一元酸,更加优选地,改性剂为四氢呋喃、乙腈、乙醇和甲醇中的一种或几种,添加剂为甲酸或乙酸。
更加优选地,改性剂的加入量为超临界co2体积的1.5~15%,添加剂加入量为超临界co2体积的0.01~0.5%。
优选地,所述步骤(3)是在避光条件下进行的,温度控制在20~30℃。
在一个优选的实施方案中,一种依托度酸光降解杂质的制备方法,包括如下步骤:
(1)、将依托度酸的样品溶解在有机溶剂中,配制成一定浓度的溶液,在溶液中加入质量浓度为1~5%的双氧水溶液,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照箱中破坏,光照强度为3000~6000lx,光照时间为10~20h得依托度酸光照破坏液;取样hplc检测。
(2)、采用超临界流体色谱法对上述依托度酸光照破坏液进行纯化,超临界流体色谱流动相分为a、b两项,a相为co2,b相为改性剂和添加剂的混合溶液,超临界流体色谱控制范围为30~35℃,色谱系统控压范围为100~300bar;采用梯度洗脱程序,收集含目标产物的洗脱流出液。
(3)、将步骤(2)所得洗脱流出液在20~30℃避光条件下快速减压浓缩蒸干即得式i光降解杂质。
优选地,步骤(1)中所述的有机溶剂为四氢呋喃、乙酸乙酯、乙腈、乙醇或甲醇;
优选地,步骤(1)中所述双氧水的加入量为依托度酸待破坏溶液体积的0.5~1.5%。
优选地,步骤(2)中所述的超临界流体色谱填料为裸硅胶、二醇基、氨基型、酰胺型或两性离子型色谱填料;更加优选地超临界流体色谱填料为裸硅胶或二醇基硅胶基质填料。
优选地,步骤(2)中所述的b相改性剂为极性非质子溶剂或c1~c4烷醇的一种或几种,添加剂为c1~c3一元酸,更加优选地,b相改性剂为四氢呋喃、乙腈、乙醇和甲醇中一种或几种,添加剂为甲酸或乙酸。
优选地,改性剂加入量为超临界co2体积的1.5~15%,添加剂加入量为超临界co2体积的0.01~0.5%。
优选地,步骤(2)所述的超临界流体色谱纯化洗脱过程为梯度洗脱,洗脱梯度表如表1所示:
表1超临界流体色谱洗脱梯度表
在另一个优选的实施方案中,一种依托度酸光降解杂质的制备方法,包括如下步骤:
(1)、将依托度酸的样品溶解在有机溶剂中,配制成一定浓度的溶液,得依托度酸待破坏液,将上述依托度酸待破坏液,置于光照箱中破坏,搅拌条件下通入空气或氧气,光照强度为3000~6000lx,光照时间为10~20h,得依托度酸光照破坏液;取样hplc检测。
(2)、采用超临界流体色谱法对上述依托度酸光照破坏液进行纯化,超临界流体色谱流动相分为a、b两项,a为co2,b相为改性剂和添加剂的混合溶液,超临界流体色谱控制范围为30~35℃,色谱系统控压范围为100~300bar;采用梯度洗脱程序,收集含目标产物的洗脱流出液。
(3)、将步骤(2)所得洗脱流出液在20~30℃避光条件下快速减压浓缩蒸干即得式i光降解杂质。
优选地,步骤(1)所述的有机溶剂为四氢呋喃、乙酸乙酯、乙腈、乙醇或甲醇;
优选地,步骤(2)所述的超临界流体色谱填料为裸硅胶、二醇基、氨基型、酰胺型或两性离子型色谱填料;更加优选地超临界流体色谱填料为裸硅胶或二醇基硅胶基质填料。
优选地,步骤(2)所述的b相改性剂为极性非质子溶剂或c1~c4烷醇的一种或几种,添加剂为c1~c3一元酸,更加优选地,b相改性剂为四氢呋喃、乙腈、乙醇和甲醇的一种或几种,添加剂为甲酸或乙酸。
优选地,改性剂加入量为超临界co2体积的1.5~15%,添加剂加入量为超临界co2体积的0.01~0.5%。
优选地,步骤(2)所述的超临界流体色谱纯化洗脱过程为梯度洗脱,洗脱梯度见表1。
本发明的另一方面在于提供一种式i化合物的检测方法,本发明所得式i依托度酸光降解杂质化合物的检测,可通过高效液相色谱检测,相对保留时间为0.95~0.97。本发明所使用的高效液相色谱仪购自美国赛默飞世尔科技。色谱条件如下:
hplc仪器型号:thermo-ultimate3000;
色谱柱:thermo─hypersil─5μm,4.6×150mm;
流动相:a相为0.6%h3po4水溶液,b相0.6%h3po4乙腈溶液;梯度洗脱。
洗脱梯度表见表2:
表2hplc检测式i杂质化合物的洗脱梯度表
检测波长:280nm
流速:1.0ml/min
柱温:30℃
本发明的技术优势在于:
(1)依托度酸光照破坏过程辅助通入氧气或空气或添加双氧水,可加强依托度酸的光降解效果,提高光降解杂质的含量,使得式ι依托度酸光降解杂质化合物的含量超过50%。
(2)使用超临界流体色谱法,采用超临界co2、改性剂和添加剂的混合物为流动相,使得式ι依托度酸光降解杂质化合物分离效率高,样品收率高。
(3)超临界流体色谱纯化的后处理方法简单快速,针对性强,有效防止样品再降解,采用本发明的技术方案,使得式ι依托度酸光降解杂质总收率超过48%,超临界流体色谱纯化收率超过90%,式ι依托度酸光降解杂质纯度高达99.01%。
附图说明
图1是实施例1依托度酸光照破坏图谱;
图2是对比实施例1依托度酸光照破坏图谱;
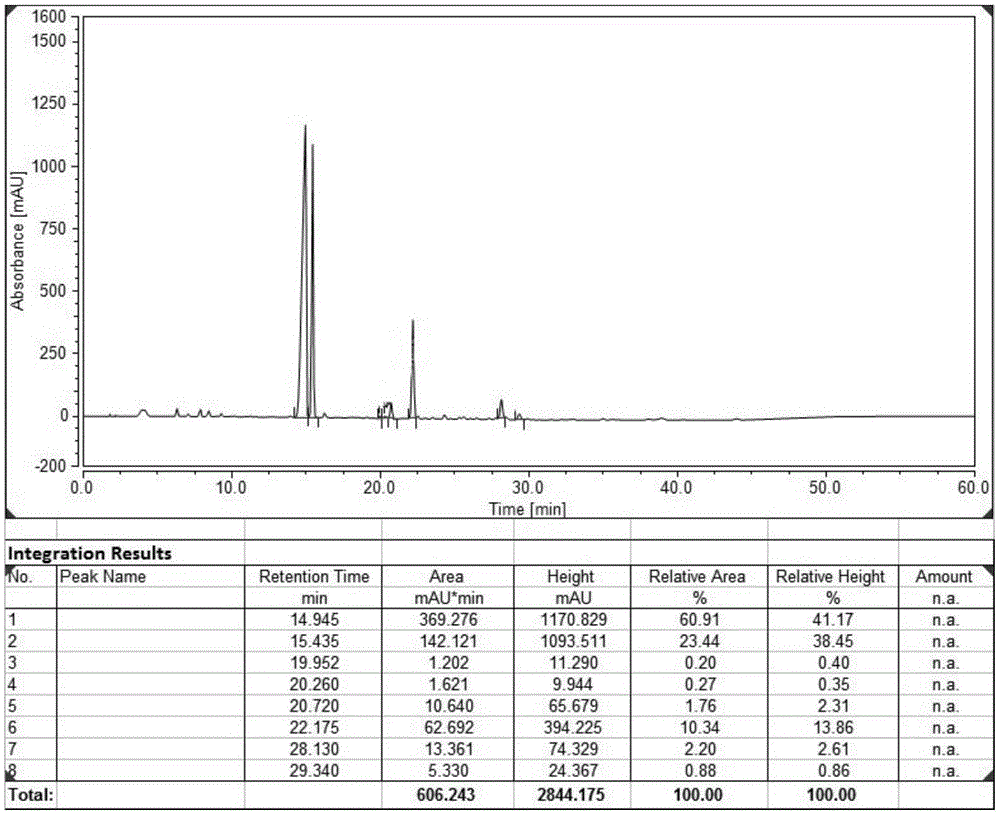
图3是实施例11依托度酸光破坏样品超临界流体色谱纯化图谱,目标产物出峰位置在保留时间为8.81处;
图4是实施例11依托度酸光降解杂质的hplc检测图谱。
具体实施方式
下面结合具体实施例对本发明做进一步详细说明,应当理解的是以下实施例仅用于例证的目的,并不是对本发明的限制,这对于本发明所述技术领域的普通技术人员而言是显而易见的。
本发明实施例中所用原料依托度酸样品由山东新时代药业有限公司提供,hplc纯度为99.06%;其他所使用的原料如无特殊说明,均为本领域常用原料,可通过商业途径获得。
实施例1:式i杂质化合物的制备
将依托度酸样品溶解在乙腈中,配制成10mg/ml的乙腈溶液100ml,加入1ml质量分数为3%的双氧水溶液,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照破坏箱,设置温度20℃,光照强度5000lx进行光照破坏15h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为60.91%。
实施例2:式i杂质化合物的制备
将依托度酸样品溶解在四氢呋喃中,配制成15mg/ml的四氢呋喃溶液100ml,加入5ml质量分数为1%的双氧水溶液,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照破坏箱,设置温度30℃,光照强度3000lx进行光照破坏20h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为57.45%。
实施例3:式i杂质化合物的制备
将依托度酸样品溶解在乙酸乙酯中,配制成10mg/ml的乙酸乙酯溶液100ml,加入0.5ml质量分数为5%的双氧水溶液,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照破坏箱,设置温度25℃,光照强度6000lx进行光照破坏10h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为55.65%。
实施例4:式i杂质化合物的制备
将依托度酸样品溶解在乙醇中,配制成20mg/ml的乙醇溶液100ml,加入1.5ml质量分数为5%的双氧水溶液,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照破坏箱,设置温度20℃,光照强度5000lx进行光照破坏15h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为53.76%。
实施例5:式i杂质化合物的制备
将依托度酸溶解在甲醇中,配制成5mg/ml的甲醇溶液,混匀得依托度酸待破坏液,将上述依托度酸置于光照破坏箱中,搅拌下不断通入空气,设置温度20℃,光照强度5000lx进行光照破坏15h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为50.89%。
实施例6:式i杂质化合物的制备
将依托度酸样品溶解在乙腈中,配制成10mg/ml的乙腈溶液100ml,加入5ml质量分数为5%的双氧水溶液,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照破坏箱,设置温度20℃,光照强度5000lx进行光照破坏15h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为16.73%。
实施例7:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为裸硅胶,颗粒大小5μm;洗脱剂为:a-co2,b为5%乙醇和0.1%甲酸;检测波长为280nm,流速为60ml/min,柱温33℃,系统背压120bar;将上述依托度酸光照破坏液上样10ml(hplc纯度60.91%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在25℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率91.4%,hplc纯度98.56%。
实施例8:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为二醇基硅胶,颗粒大小5μm;洗脱剂为:a-co2,b为10%乙腈和0.01%乙酸;检测波长为280nm,流速为60ml/min,柱温30℃,系统背压300bar;将上述依托度酸光照破坏液上样10ml(hplc纯度57.45%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在20℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率92.3%,hplc纯度98.86%。
实施例9:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为氨基型硅胶,颗粒大小10μm;洗脱剂为:a-co2,b为1.5%四氢呋喃和0.3%甲酸;检测波长为280nm,流速为60ml/min,柱温35℃,系统背压200bar;将上述依托度酸光照破坏液上样10ml(hplc纯度53.76%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在30℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率90.1%,hplc纯度98.54%。
实施例10:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为酰胺基硅胶,颗粒大小5μm;洗脱剂为:a-co2,b为15%甲醇和0.5%乙酸;检测波长为280nm,流速为60ml/min,柱温33℃,系统背压150bar;将上述依托度酸光照破坏液上样10ml(hplc纯度60.91%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在25℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率91.6%,hplc纯度98.02%。
实施例11:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为二醇基硅胶,颗粒大小5μm;洗脱剂为:a-co2,b为10%甲醇和0.1%乙酸;检测波长为280nm,流速为60ml/min,柱温33℃,系统背压120bar;将上述依托度酸光照破坏液上样10ml(hplc纯度60.91%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在25℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率94.5%,hplc纯度99.01%。
实施例12:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为裸硅胶,颗粒大小10μm;洗脱剂为:a-co2,b为10%甲醇;检测波长为280nm,流速为60ml/min,柱温33℃,系统背压150bar;将上述依托度酸光照破坏液上样10ml(hplc纯度60.91%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在25℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率72.8%,hplc纯度96.33%。
实施例13:式i杂质化合物的纯化
将依托度酸光照破坏液采用超临界流体色谱纯化,色谱柱尺寸为19mm×150mm,填料为两性离子型色谱填料,颗粒大小10μm;洗脱剂为:a-co2,b为10%异丙醇和0.1%甲酸;检测波长为280nm,流速为60ml/min,柱温33℃,系统背压120bar;将上述依托度酸光照破坏液上样10ml(hplc纯度60.91%),运行表1的洗脱梯度表,收集含目标产物的洗脱流出液,取样检测;将上述洗脱流出液在25℃水浴条件下,避光快速减压浓缩至干,得式i杂质化合物,纯化收率80.6%,hplc纯度97.76%。
对比实施例1:式i杂质化合物的制备
将依托度酸样品溶解在乙腈中,配制成10mg/ml的乙腈溶液100ml,混匀得依托度酸待破坏液;将上述依托度酸待破坏液放置于光照破坏箱,设置温度20℃,光照强度5000lx进行光照破坏15h,得依托度酸光照破坏液;取样进行hplc检测,式i杂质化合物的hplc纯度为5.88%。
对比实施例2:式i杂质化合物的纯化
使用反相高效液相色谱法,选择使用
表3反相高效液相色谱洗脱梯度表
制备所得式i依托度酸光降解杂质化合物的结构确证:
hresi-ms(m/z):320.2069[m+h]+;
1hnmr(500mhz,cdcl3)δh:7.98(s,nh),7.49(d,j=7.7hz,1h),7.39(t,j=7.6hz,1h),7.25(d,j=7.4hz,1h),2.77(dq,j=7.5,1.0hz,2h),1.26(t,j=7.5hz,3h),4.45(t,j=12.0hz,1h),4.16(ddd,j=13.5,4.7,2.1hz,1h),3.17(m,1h),2.47(dd,j=14.7,3.4hz,1h),3.18(d,j=14.6hz,1h),2.86(d,j=14.6hz,1h),2.11(dq,j=14.4,7.3hz,1h),1.96(dq,j=12.7,6.4hz,1h),1.13(t,j=7.5hz,3h).
13cnmr(125mhz,cdcl3)δc:204.6(c),177.5(c),172.0(c),142.8(c),138.9(c),133.5(c),132.1(ch),128.7(ch),124.7(ch),80.7(c),63.7(ch2),42.9(ch2),35.9(ch2),30.5(ch2),23.6(ch2),14.2(ch3),7.9(ch3).
- 还没有人留言评论。精彩留言会获得点赞!