一种脲基嘧啶酮前体的制备方法与流程
本发明属于超分子材料中间体的合成领域,具体涉及一种脲基嘧啶酮前体的合成工艺。
背景技术:
自组装在自然界中普遍存在,特别是诸如dna和蛋白质的生物系统中,已经被广泛应用于通过不同的非共价相互作用的组合来构建高度复杂和明确定义的纳米结构,由于氢键有着方向性、选择性和可逆性,使其成为超分子聚合物中最常用的非共价键连接。1997年,荷兰科学家meijer教授等首次成功合成了脲基嘧啶酮(upy)基元,也是目前超分子化学领域应用最广泛的基元(science,1997,278,1601-1604.)。该基元的一种异构体可以通过自互补型四重氢键作用自络合为二聚体。一般来说,upy的制备由其前体接上脲基而成,得到的upy具有超高的自络合常数(chcl3中kass>107m-1,),利用upy基元构筑一系列新型的超分子聚合物受到众多科学家的极大关注。
从目前的研究工作来看,合成upy前体的种类繁多,且合成路线各有不同。本发明提供一种明晰的且收率高的合成upy前体的改进方法。
技术实现要素:
本发明的目的旨在提供一种脲基嘧啶酮前体的合成方法
所述脲基嘧啶酮前体记为化合物d,其化学结构式如下:
所述脲基嘧啶酮前体的合成路线如下:
所述脲基嘧啶酮前体的合成方法,包括以下步骤:
1)丙二酸二乙酯钾盐(记为化合物a)的合成:
向反应容器中加入丙二酸二乙酯和甲醇混合搅拌,将koh加入混合液中,室温搅拌超过2h,停止搅拌,加热回流反应至白色悬浮液变成澄清溶液,抽滤得到白色晶体,在真空干燥箱干燥得到化合物a。
2)2-乙基己酰氯(记为化合物b)的合成:
向反应容器中加入2-乙基己酸与氯化亚砜,室温下过夜搅拌,停止搅拌,反应液旋转蒸发除去大部分socl2,向容器加入甲苯继续旋转蒸发除去残留的甲苯和socl2,得到化合物b。
3)β-酮酸酯(记为化合物c)的合成:
在反应容器中加入化合物a,n2保护下加入无水乙腈,搅拌并用冰盐浴冷却,加入碳酸胍,用无水乙腈润洗,继续搅拌,向反应液中加入无水mgcl2、无水乙腈搅拌,撤去冰盐浴,室温搅拌,在冰盐浴条件下,逐滴加入化合物b,并用无水乙腈润洗,继续搅拌,将冰浴改为油浴,旋蒸除去乙腈得到黄色固体,加入hcl直至无气体放出,加入乙醚洗涤和萃取,收集油相,再用饱和nahco3水溶液洗涤除去hcl,用无水mgso4干燥,旋蒸得到化合物c。
4)脲基嘧啶酮前体(记为化合物d)的合成:
在反应容器中加入β-酮酸酯、碳酸胍和无水乙醇,加热回流反应,停止加热,冷却至室温,过滤得黄色液体,旋蒸除去乙醇,加入chcl3互溶,用饱和nahco3水溶液洗涤,收集有机相,用无水mgso4干燥,过滤,旋蒸得到黄色油状物,加入大量正己烷,有淡黄色固体析出,抽滤,在真空干箱干燥得到化合物d。
在步骤1)中,所述丙二酸二乙酯与koh的摩尔比为1:1;所述koh加入混合液中的具体操作为:先将koh溶于甲醇中,再滴加入混合液中;所述加热回流反应为在100℃下反应1~2h。
在步骤2)中,所述2-乙基己酸与氯化亚砜的摩尔比为1:2.5;所述旋转蒸发设定的温度为76~80℃。
在步骤3)中,所述n2保护可连续抽真空后重氮气循环2~4次;所述碳酸胍需先用koh干燥;所述油浴温度设定为32℃;所述hcl应为3mol/l;所述乙醚洗涤和萃取应分3~4次进行;所述饱和nahco3水溶液洗涤应分1~2次进行。
在步骤4)中,所述无水乙醇需用分子筛提前干燥,所述加热回流反应可在110~120℃下反应2d;所述旋转除去乙醇温度可设定为50~60℃;所述chcl3为水洗15~18次干燥处理后的chcl3;所述饱和nahco3水溶液洗涤应分2~3次进行;所述干燥的条件可在真空50℃下干燥2h。
本发明改进了一种upy前体衍生物,该化合物在upy前体上连接了一个烷基链,大大增加了溶解度,从而为upy的合成提供了极大便捷。
本发明的优点如下:
(1)本合成方法选用毒性低、廉价易得的物质作为原料,在合成丙二酸二乙酯钾盐时采用甲醇作为溶剂,甲醇对koh的溶解性更好,且钾盐的产率有显著提升。
(2)本合成方法在合成2-乙基己酰氯时采用甲苯作为共沸溶剂,与苯相比,甲苯的毒性很低,且所得产率更高。
(3)本合成方法合成的脲基嘧啶酮前体,在upy前体上连接了一条烷基链,增加了溶解度。
附图说明
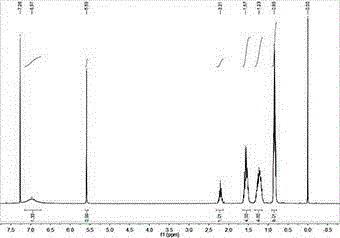
图1为化合物d在cdcl3中的核磁共振氢(1hnmr)谱图,其中横坐标为化学位移(ppm)。
具体实施方式
为了更好的理解本发明,下面通过具体的实施例对本发明作进一步说明
实施例1:
步骤1、化合物a的合成
向250ml圆底烧瓶中加入10g丙二酸二乙酯和40ml甲醇混合搅拌,将3.65gkoh用60ml甲醇溶解,缓慢滴加入混合液中,在室温下搅拌超过2h,停止搅拌,加热回流反应至白色悬浮液变成澄清溶液,加冰浴冷却至室温,成白色絮状物,抽滤得到白色晶体,在真空干燥箱干燥得到化合物a,产率为88%。
步骤2、化合物b的合成
在100ml三口烧瓶上接上冷凝管,并在冷凝管口加一干燥管,后接尾气吸收naoh溶液,向三口烧瓶中加入26.5ml2-乙基己酸和24ml氯化亚砜,在室温下搅拌,移除尾气装置,过夜搅拌,将反应液移入250ml蒸馏烧瓶中,旋转蒸发仪的温度设为76℃,真空旋转蒸发除去大部分socl2,向蒸馏烧瓶中加入20ml甲苯共沸,继续旋转蒸发除去残留的甲苯和socl2,得到微黄色液体,即化合物b。
步骤3、化合物c的合成
向500ml三口烧瓶中加入24.4g化合物a,连续2~4次抽真空,充氮气的循环,排出体系中的氧气和水分。在n2保护下加入135ml无水乙腈,搅拌并用冰盐浴冷却,加入31.5ml干燥过的碳酸胍,用15ml无水乙腈润洗,继续搅拌,向反应液中加入16.5g无水mgcl2、50ml无水乙腈搅拌,得到白色絮状物,撤去冰盐浴,室温搅拌18h,在冰盐浴条件下,逐滴加入11g化合物b,并用10ml无水乙腈润洗,继续搅拌,将冰浴改为油浴,温度设定为32℃,加热1d后,将反应液移入蒸馏烧瓶,旋转蒸发仪的温度设为50℃,真空旋蒸除去乙腈得到黄色固体,向蒸馏烧瓶中加入150ml3mol/l的hcl直至无气体放出,液体转移至分液漏斗中,加入乙醚(50ml×4)洗涤和萃取,收集上层油相,再用饱和nahco3水溶液(50ml×2)洗涤除去hcl,用无水mgso4干燥,旋转蒸发仪的温度设为30℃,真空旋蒸得到化合物c。
步骤4、化合物d的合成
向500ml三口烧瓶中加入13gβ-酮酸酯、32.79g碳酸胍、300ml经分子筛干燥的无水乙醇,在114℃下加热回流反应2d,停止加热,冷却至室温,溶液分层,上层为黄色溶液,下层为白色沉淀,过滤得黄色液体,旋转蒸发仪的温度设为50℃,真空旋蒸除去乙醇,加入300mlchcl3互溶,用饱和nahco3水溶液(100ml×2)洗涤,收集下层白色溶液,用无水mgso4干燥,过滤,滤液移至蒸馏烧瓶,旋转蒸发仪温度设为20℃,真空旋蒸得到黄色油状物,加入大量正己烷,有淡黄色固体析出,抽滤得到白色固体,在真空干箱干燥得到化合物d。
实施例2~5:
化合物a、化合物b和化合物c的合成同实施例1,化合物d的合成参照实施例1的步骤4进行合成,仅改变β-酮酸酯与碳酸胍摩尔比为1:2.0、1:2.5、1:3.5、1:4.0,化合物d的产率依次为58%、65%、70%、64%,优选β-酮酸酯与碳酸胍摩尔比为1:3.5。
上述实施例仅用来进一步说明本发明的一种给脲基嘧啶酮前体的合成方法,但本发明并不局限于实施例,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其他不同形式的变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变动仍处于本发明的保护范围之列。
- 还没有人留言评论。精彩留言会获得点赞!
- 5-(4-氟苯基)-N–(4-氯苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(3-(二甲基氨基)丙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 一种5-芳基吡唑并[1,5-a]嘧啶衍生物的合成方法
- 5-(4-氟苯基)-N,N-二甲基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 一类含呋喃的嘧啶联吡唑甲酰胺类衍生物及其制备方法和用图
- 5-(4-氟苯基)-7-三氟甲基吡唑并[1,5-a] 嘧啶-3-羧酸乙酯的合成方法
- 5-(4-氟苯基)-7-三氟甲基吡唑并[1,5-a] 嘧啶-3-羧酸的合成方法
- 5-(4-氟苯基)-N–1-(3-羟基金刚烷基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 作为人中性白细胞弹性蛋白酶抑制剂的四氢三唑并嘧啶衍生物的制作方法