2,5-二烷基呋喃的一步合成法的制作方法
本发明属于有机合成领域,具体涉及一种2,5-二烷基呋喃一步合成方法。
背景技术:
2,5-二烷基呋喃作为一种重要的呋喃衍生物,广泛地分布在人们的生活当中,比如,加热的咖啡、烘焙的食品、调味料等都可以发现2,5-二烷基呋喃的存在。2,5-二烷基呋喃也可以作为美容产品中的祛斑成分被人们用于美容治疗过程,同时人们也将它作为中间原料合成高效的抗病毒医药试剂。此外,2,5-二烷基呋喃分子含有刚性的五元环结构和α位的烷基链,使之可以经过氧化还原得到呋喃二酸和呋喃二醇,这些二次合成的产物可直接用于高性能聚酯、环氧树脂、聚酰胺、聚氨酯等高性能工程塑料的制备,在化工行业中占据着举足轻重的作用。
2,5-二烷基呋喃虽然在生活中分布广泛,但是分布较为分散且含量较少,自然提取显然不利于其大规模利用。同时生物酶发酵也是提取2,5-二烷基呋喃的一种途径,但是高昂的技术成本以及高精确度的生物繁殖技术同样也不利于其大规模生产。如今,化学合成2,5-二烷基呋喃是最有希望使其大规模应用的途径。迄今为止,2,5-二烷基呋喃的合成主要由两种方法已被报道。
第一种合成方法是首先将呋喃与马来酸酐通过狄尔斯-阿尔德反应(diels-alderreaction)制备2,3-二甲酸酐-7-氧杂二环[2.2.1]庚-5-烯。然后将2,3-二甲酸酐-7-氧杂二环[2.2.1]庚-5-烯与碘代烃在酸性催化剂作用下通过傅列德尔-克拉夫茨烷基化反应(friedel-craftsalkylation)反应制备2,5-二烷基呋喃[1]。
以上反应方法虽然可以成功的制备2,5-二烷基呋喃,且保持收率在60%以上。但是合成过程涉及到质子性酸性催化剂的使用,如磷酸、硝酸、硫酸、盐酸等,这样势必会为后处理中和过量的酸性催化剂增加工作量。同时酸性催化剂的使用也会对金属管路造成一定的腐蚀,需要增加对金属设备保养和维护频率,甚至降低金属设备的使用寿命。这种不符合可持续发展绿色经济的合成方法显然不利于2,5-二烷基呋喃的工业化生产及推广。更为甚者,反应过程需要采用具有高价格、高挥发性、毒性的碘代烃,虽然有利于产率的提高,但是也为生产操作技术人员的生命安全减小了保障的砝码。最后,此种合成方法需要运行周期大约在30h左右,较长的合成周期也是限制其应用的短板。
第二种合成方法是将将呋喃与碘代烃在酸性催化剂存在下,直接通过傅列德尔-克拉夫茨烷基化反应(friedel-craftsalkylation)合成2,5-二烷基呋喃。
新改进的合成方法较第一种简化了反应过程,但是合成过程仍然需要借助质子性酸性催化剂,如磷酸、硝酸、硫酸、盐酸等,这样同样不可避免地为后处理中和过量的酸性催化剂增加工作量。同时酸性催化剂的使用也会对金属管路造成不可规避的腐蚀,最终需要增加对金属设备的保养和维护频率,甚至降低金属设备的使用寿命。这种不符合持续发展绿色经济的合成方法显然不利于2,5-二烷基呋喃的工业化生产及推广。更为甚者,反应过程同样需要采用具有高价格、高挥发性、毒性的碘代烃,虽然有利于产率的提高,但是也为生产操作技术人员的生命安全减小了保障的砝码。最后,此种合成方法需要运行周期大约在20h左右,较长的合成周期也是限制其应用的短板。
因此,寻找一种绿色环保经济高效的2,5-二烷基呋喃的合成方法很有必要。
技术实现要素:
本发明的目的在于提出一种用于合成2,5-二烷基呋喃的绿色环保经济高效一步合成法。
基于上述目的,本发明采取如下技术方案:
2,5-二烷基呋喃的一步合成法,合成路线如下:
r为2至16个碳原子的饱和链烃,具体步骤如下:
(1)将呋喃、n,n'-四甲基乙二胺(tmeda)和溶剂四氢呋喃(thf)加入反应瓶中,-5~0℃搅拌15min~30min;
(2)滴加正丁基锂(n-buli)正己烷溶液,加毕,-5~0℃下继续搅拌15min~30min;
(3)混合物升温至60~70℃回流,并维持4-5h;
(4)混合物移至室温冷却,再在-5~0℃下搅拌15min~30min;
(5)滴加溴代烷烃,加毕,升温至室温并搅拌至反应完全,经后处理即可得到2,5-二烷基呋喃。
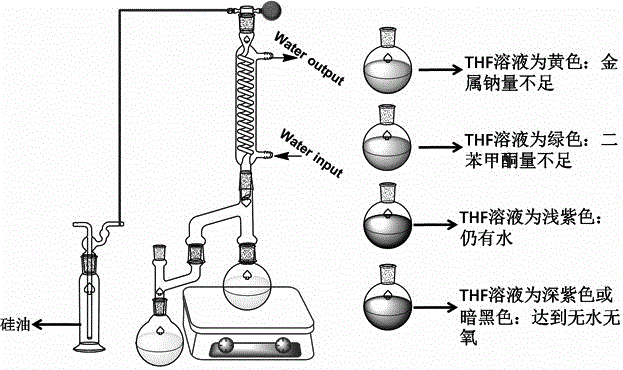
进一步地,反应过程所选溶剂thf需经钠干燥,二苯甲酮显色,颜色为深紫色时,常压蒸馏储存于安培瓶中备用。
进一步地,反应过程需要在氮气保护下进行。
进一步地,反应过程所选原料呋喃、tmeda、n-buli和溴代烷烃的摩尔比为5:9:9:9。
进一步地,反应后处理是指将饱和氯化铵水溶液加入反应完后的混合物中,用有机溶剂进行萃取,合并有机相并用无水硫酸钠干燥,减压旋出溶剂,最后经减压蒸馏得到2,5-二烷基呋喃。后处理所用有机溶剂为二氯甲烷、氯仿、乙酸乙酯或乙醚。
进一步地,反应原料溴代烷烃中烷基r可选自下述结构中的一种:
进一步地,反应过程所需要温度(-5~0℃)可通过冰水浴来实现。
进一步地,本发明所述合成方法可实现绿色环保无废处理的高效合成,具体为后处理所用无机盐为氯化铵。后处理的水相含有氯化铵和溴化锂,混合物经过高温处理,使氯化铵分解可获得化学纯溴化锂,用于吸收式制冷剂,氯化氢脱除剂、纤维蓬松剂、催眠剂和镇静剂,还用于感光工业、分析化学试剂以及某些高能电池中的电解质,实现反应的无废循环经济。
本发明具有下述优点:
1)反应过程中加入tmeda可以与正丁基锂反应生成n,n'-四甲基乙二胺基锂,减弱了亲核试剂的活性,避免副反应和副产物的产生;
2)反应过程中所需温度保持在-5-0℃范围内,一般的冰水浴足以满足要求;
3)反应中未完全反应的呋喃原料可以通过旋转蒸发收集,循环利用率高;
4)反应目标产物2,5-二烷基呋喃可通过减压蒸馏获得,减压蒸馏可以通过循环水泵获得,降低了合成了对设备的要求,节约了使用油泵的成本;
5)反应过程不使用高价格、高挥发性、高毒性原料,反应后处理简单易行,整体绿色环保,可实现高效率原子经济合成,有利于工业化生产及效率的最大化。
附图说明
图1是本发明蒸馏纯化四氢呋喃的装置及颜色变化示意图;
图2是本发明2,5-二烷基呋喃的合成装置示意图;
图3是实施例3中2,5-二异辛基呋喃的核磁共振氢谱(1h-nmr)。
具体实施方式
为了使本发明的技术目的、技术方案和有益效果更加清楚,下面结合附图和具体实施例对本发明的技术方案作出进一步的说明。
实施例1
2,5-二乙基呋喃的合成:
如图2所示,将呋喃(50mmol,3.404g),n,n'-四甲基乙二胺(90mmol,10.459g)和100ml无水无氧四氢呋喃(无水无氧四氢呋喃的重蒸过程详见图1,为本领域技术人员公知的方法,此处不再详述)加入到装有恒压滴液漏斗和回流冷凝管的250ml三口烧瓶中,微抽真空,充氮气,依次往复3次。烧瓶置于冰水浴中搅拌0.5h,开始缓慢滴加正丁基锂的正己烷溶液(90mmol,2.5mol/l,36ml),加毕,冰水浴下继续搅拌0.5h。紧接着将混合物移至室温,并升温至66℃回流且保持4h。将混合物冷却至室温,然后移至冰水浴中搅拌0.5h,开始缓慢滴加溴代乙烷(90mmol,9.807g),加毕,移至室温搅拌过夜。反应混合物加入到100ml的饱和氯化铵水溶液中,用二氯甲烷(3×50ml)萃取。合并有机相,并用无水硫酸钠干燥,过滤,收集有机相并经减压旋除溶剂。最后粗产物经减压蒸馏得到最终目标产物,为无色液体(4.368,70.4%),纯度98%。
1h-nmr(cdcl3,400hz,ppm)δ:5.98(d,j=3.6hz,1h),2.44(m,2h),1.26(t,j=4.0hz,3h)
实施例2
2,5-二辛基呋喃的合成:
将呋喃(50mmol,3.404g),n,n'-四甲基乙二胺(90mmol,10.459g)和100ml无水无氧四氢呋喃加入到装有恒压滴液漏斗和回流冷凝管的250ml三口烧瓶中,微抽真空,充氮气,依次往复3次,然后置于冰水浴中搅拌0.5h。缓慢滴加正丁基锂的正己烷溶液(90mmol,2.5mol/l,36ml),加毕,冰水浴下继续搅拌0.5h。然后将混合物移至室温,并升温至66℃回流且保持4h。紧接着混合物冷却到室温,然后移至冰水浴中搅拌0.5h。缓慢滴加溴代辛烷(90mmol,17.381g),加毕,移至室温搅拌过夜。加入到100ml的饱和氯化铵水溶液中,用二氯甲烷(3×50ml)萃取。合并有机相,用无水硫酸钠干燥,过滤并收集有机相,经减压旋除溶剂。最后粗产物经减压蒸馏得到目标产物,为无色液体(10.741,73.5%),纯度99%。
1h-nmr(cdcl3,400hz,ppm)δ:5.95(d,j=3.6hz,1h),2.42(t,j=6.4hz,2h),1.28(m,12h),0.88(t,j=3.6hz,3h)
实施例3
2,5-二异辛基呋喃的合成:
将呋喃(50mmol,3.404g),n,n'-四甲基乙二胺(90mmol,10.459g)和100ml无水无氧四氢呋喃加入到装有恒压滴液漏斗和回流冷凝管的250ml三口烧瓶中,微抽真空,充氮气,依次往复3次,然后置于冰水浴中搅拌0.5h。缓慢滴加正丁基锂的正己烷溶液(90mmol,2.5mol/l,36ml),加毕,冰水浴下继续搅拌0.5h。然后将混合物移至室温,并升温至66℃回流且保持4h。紧接着混合物冷却到室温,然后置于冰水浴中搅拌0.5h。缓慢滴加溴代异辛烷(90mmol,17.381g),加毕,移至室温搅拌过夜。加入到100ml的饱和氯化铵水溶液中,用二氯甲烷(3×50ml)萃取。合并有机相,用无水硫酸钠干燥,过滤并收集有机相,经减压旋除溶剂。最后粗产物经减压蒸馏得到目标产物,为无色液体(11.633,79.6%),纯度98%。
化合物的1h-nmr如图3所示,1h-nmr(cdcl3,400hz,ppm)δ:5.99(d,j=4.0hz,1h),2.59(d,j=4.0hz,2h),1.68(m,1h),1.34(m,8h),0.91(t,j=3.2hz,6h)。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!