一种他替瑞林的合成方法与流程
本发明涉及一种具有改善脊髓小脑变性患者的运动失调的医药产品的合成方法,特别是一种他替瑞林的合成方法。
背景技术:
他替瑞林,其结构式如下:
他替瑞林产品与trh类似,其内分泌作用比trh弱,但是在体内比trh更加稳定。他替瑞林产品由日本田边三菱制药株式会社研制成功,2000年9月在日本首次上市,目前在中国尚未上市及销售。目前文献报道他替瑞林主要有几条合成路线,但是都未能避开形成二酮哌嗪(dkp)的路径,而是通过控制反应条件来降低该杂质。中国公开专利文献cn85105655a公开的方法为甲基乳清酸与l-组氨酸-l-脯氨酸的先导部分反应,然后用催化剂催化脱除保护基获得产品。中国公开专利文献cn103588862a和cn85105655a共同公开了另外一种制备他替瑞林的方法,甲基乳清酸与l-组氨酸-l-脯氨酸的先导部分反应,然后25%的胺甲醇溶液脱除保护基获得产品。其中中国公开专利文献cn103588862a虽然将乳清酸的合成方法叠加到了他替瑞林的合成方法内,但本质上并未对他替瑞林的制备方法进行创新。文献《bulletinofthechemicalsocietyofjapan》1971年第44期,1689-1691页报道的方法,采用甲基乳清酸与l-组氨酸-l-脯氨酸的先导部分缩合反应,然后脱保护获得产品。该路线不仅没有避开形成二酮哌嗪的核心步骤,且用到了氯甲酸甲酯、碘甲烷等剧毒物质,hbr等强酸物质,对环境和操作人员及其不好,且由于反应条件比较苛刻,不易产业化。
技术实现要素:
本发明所要解决的技术问题是针对现有技术路线中二酮哌嗪存在的问题,提供了一种新的他替瑞林的合成方法,该方法避开了产生二酮哌嗪的反应过程,降低杂质,有效提高收率和产品品质。
本发明所要解决的技术问题是通过以下的技术方案来实现的。本发明是一种他替瑞林的合成方法,其特点是:该方法以2-氰基吡咯烷为起始物料与氨基和侧链保护的组氨酸进行缩合,形成酰胺键,再与1-甲基-4,5-二氢乳清酸缩合,最后通过氧化和脱保护,获得他替瑞林。
本发明方法的合成路线如下:
本发明所述的一种他替瑞林的合成方法,其进一步优选的技术方案是:
1、缩合反应通过硫苄酯转酯反应完成,或者通过混合酸酐完成缩合反应,或者通过活化酯反应完成缩合反应,或者采用催化剂缩合反应,或者采用酰氯缩合反应。
2、氨基保护基选自boc、z、z-cl、fmoc或h。
3、组氨酸侧链保护基选自boc、z、bzl、trt、tos、bom、dnp或h。
4、1-甲基-4,5-二氢乳清酸中3号位氮保护基选自boc、z、z-cl、bzl、trt、tos、bom、dnp、fmoc或h。
本发明方法以2-氰基吡咯烷为起始物料,与氨基和侧链保护的组氨酸进行缩合,形成酰胺键,有效避免了dkp的形成。再采用室温下dcc、hobt与1-甲基-4,5-二氢乳清酸缩合,最后通过氧化和脱保护,获得他替瑞林产品。
与现有技术相比,本发明方法具有以下有益效果:该方法经过缩合、氧化水解、脱保护等步骤获得高纯度的他替瑞林产品,其方法步骤完全避免了常规方法形成二酮哌嗪(dkp)的步骤,使得产品杂质少,易纯化,收率高,溶液易于回收套用。同时,本方法还缩短了合成周期、避免了传统方法中苛刻的反应条件,而且收率高、产品纯度好、易于纯化、成本低、反应条件温和、适合于工业化生产。
附图说明
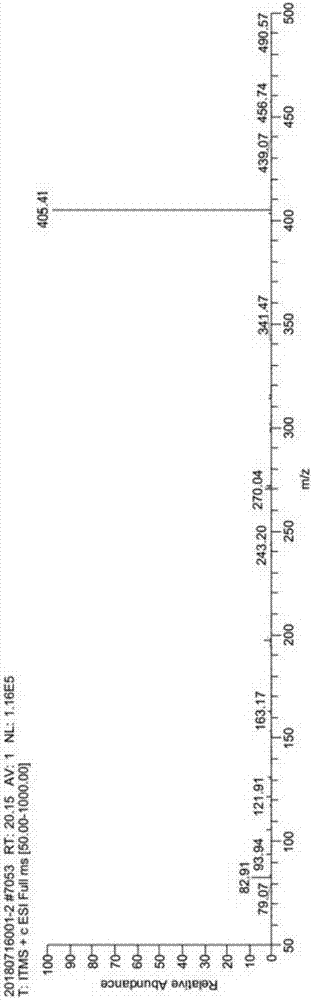
图1为实施例合成得到的他替瑞林产品的质谱图。
具体实施方式
实施例1,一种他替瑞林的合成方法:该方法以2-氰基吡咯烷为起始物料与氨基和侧链保护的组氨酸进行缩合,形成酰胺键,再与1-甲基-4,5-二氢乳清酸缩合,最后通过氧化和脱保护,获得他替瑞林。
缩合反应通过硫苄酯转酯反应完成,或者通过混合酸酐完成缩合反应,或者通过活化酯反应完成缩合反应,或者采用催化剂缩合反应,或者采用酰氯缩合反应。
氨基保护基选自boc、z、z-cl、fmoc或h。
组氨酸侧链保护基选自boc、z、bzl、trt、tos、bom、dnp或h。
1-甲基-4,5-二氢乳清酸中3号位氮保护基选自boc、z、z-cl、bzl、trt、tos、bom、dnp、fmoc或h。
实施例2,他替瑞林合成实验:
1、(9h-芴-9-基)甲基((s)-1-((s)-2-氰基吡咯烷-1-基)-1-氧代-3-(1-三苯甲基-1h-咪唑-5-基)丙-2-基)氨基甲酸(化合物1)的合成
溶液1:称取5毫摩的fmoc-his(trt)-oh和6毫摩的hobt,置于100ml的圆底烧瓶中,加入10mldmf溶解,磁力搅拌下冷却至0-5度备用。
溶液2:称取6毫摩的dcc,置于小烧杯中,用3ml的dmf溶解,冷却至0-5度备用
溶液3:称取5毫摩的2-氰基吡咯烷、置于小烧杯中,用3ml的dmf溶解,冷却至0-5度备用。
将溶液2加入到溶液1中,0-5度下反应30分钟,25-30度下,反应2小时,tlc跟踪至反应结束,过滤除去不溶固体dcu,将滤液冷却至0-5度,搅拌下,慢慢滴加溶液3,滴加完成后反应30分钟,升温至25-30度下反应2小时,tlc确定反应结束(具体反应时间以tlc时间为准)。
反应结束后,0-5度下,慢慢滴加150ml稀盐酸溶液,搅拌下,出现大量白色沉淀,过滤得到固体。固体用400ml乙酸乙酯溶解,洗涤液进行洗涤。洗涤液分别为稀碳酸氢钠50ml×3,稀盐酸30ml×3,最后用饱和食盐水洗涤50ml×3,溶液转移至三角瓶,加入无水硫酸钠或者无水硫酸镁进行干燥。干燥约10小时。减压蒸馏除去溶剂(回收利用),得到白色固体。hplc纯度为98.6%,收率为97.5%。
2、(s)-1-(n-对三苯甲基-l-组氨酰基)吡咯烷-2-甲腈(化合物2)的合成
称取5毫摩的化合物1,置于100ml的圆底烧瓶中,加入含30%哌啶的15mldmf溶液,磁力搅拌下20-25度反应2小时。tlc确定反应结束。加入100ml冰水,产生大量白色沉淀,过滤得到白色沉淀,用异丙醚(50ml×3)室温打浆洗涤除去保护剂残留物,获得白色固体产品。hplc纯度为99.2%,收率为98.4%。
3、(s)-n-((s)-1-((s)-2-氰基吡咯烷-1-基)-1-氧代-3-(1-三苯甲基-1h-咪唑-5-基)丙-2-基)-1-甲基-2,6-二氧代六氢嘧啶-4-甲酰胺(化合物3)
溶液1:称取5毫摩的1-甲基-4,5-二氢乳清酸和6毫摩的hobt,置于100ml的圆底烧瓶中,加入10mldmf溶解,磁力搅拌下冷却至0-5度备用。
溶液2:称取6毫摩的dcc,置于小烧杯中,用3ml的dmf溶解,冷却至0-5度备用
溶液3:称取5毫摩的化合物2,置于小烧杯中,用10ml的dmf溶解,冷却至0-5度备用。
将溶液2加入到溶液1中,0-5度下反应30分钟,25-30度下,反应2小时,tlc跟踪至反应结束,过滤除去不溶固体dcu,将滤液冷却至0-5度,搅拌下,慢慢滴加溶液3,滴加完成后反应30分钟,升温至25-30度下反应2小时,tlc确定反应结束(具体反应时间以点板时间为准)。
反应结束后,0-5度下,慢慢滴加150ml稀盐酸溶液,搅拌下,出现大量白色沉淀,过滤得到固体。固体用400ml乙酸乙酯溶解,分液漏斗进行洗涤。分别为稀碳酸氢钠50ml×3,稀盐酸30ml×3,最后用饱和食盐水洗涤50ml×3,溶液转移至三角瓶,加入无水硫酸钠或者无水硫酸镁进行干燥。干燥约10小时。减压蒸馏除去溶剂(回收利用),得到白色固体。hplc纯度为98.3%,收率为96.7%。
4、(s)-n-((s)-1-((s)-2-氨基甲酰基吡咯-1-基)-1-氧代-3-(1-三苯甲基-1h-咪唑-5-基)丙-2-基)-1-甲基-2,6-二氧代六氢嘧啶-4-甲酰胺(化合物4)
称取5毫摩的化合物3,溶于150ml的dmso中,加入30%的双氧水和碳酸钾溶液60ml,室温下反应10分钟结束,获得化合物4。
5、他替瑞林的合成
将化合物4溶于30ml含5%tfa的dcm溶液中,室温反应30分钟,反应结束。加入冰异丙醚150ml,析出大量白色固体,白色固体为他替瑞林(粗品)。白色固体用制备液相进行纯化,收率为90.2%,hplc纯度为99.67%。ms:405.41;
产图质谱图参照图1。
- 还没有人留言评论。精彩留言会获得点赞!