一类生物亲和探针分子及其制备方法与应用与流程
本发明属于化学医药技术领域,涉及一类生物亲和探针分子及其制备方法与应用。
背景技术:
天然(陆地生物,海洋生物)来源的活性化合物往往具有奇特的结构和强力的生物活性,是活性先导化合物的重要来源。在新药开发的过程中,发现具有活性的先导化合物仅是前提,确定活性化合物的靶标也具有十分重要的意义。活性化合物靶标的明确不仅对活性化合物进一步的结构优化具有指导意义,而且对研究其在生命体内的作用机制也是关键。
基于天然活性化合物全合成技术的发展及蛋白质质谱等分析手段的进步,近十余年,利用小分子探针钓取活性化合物的靶标蛋白,并深入研究其在体内信号转导等过程中的作用机制,解明生命现象,指导新药开发等已成为一个新的热点领域。
一般活性小分子探针是将活性化合物与作为分离基团的生物素(biotin)等之间通过一定长度的连接基团连接起来,合成得到小分子探针。利用生物素与链霉亲和素的结合作用而富集纯化靶标蛋白,可是由于活性小分子中导入了连接集团和分离基团,分子尺寸变大,降低了小分子同靶标蛋白的结合能力,除非活性小分子与靶标的结合作用极强或者靶标蛋白在细胞中高浓度存在,大多数小分子探针与蛋白之间的结合作用都会很弱。因此设计合成新型生物亲和探针,提高小分子探针与蛋白之间的结合作用就具有重要意义。
为了提高这种结合作用,研究者们将叠氮基团、三氟甲基苯基双吖丙啶与二苯甲酮类光反应亲和基团导入到探针分子中,利用光照条件下,光亲和基团能够与蛋白表面的氨基酸残基形成不可逆的共价结合。确实能提高探针分子与靶标的结合作用。可是由于光反应基团的反应性强,选择性低,能先于活性小分子与靶标蛋白之间的特异性结合之前,与非靶标蛋白、溶剂分子以及非蛋白类化合物反应,使探针分子与靶标蛋白的结合效果并没有像期待的那样大幅提高;同时由于光亲和基团与蛋白(靶标蛋白与非靶标蛋白)表面的氨基酸残基形成不可逆的共价结合,给后续的蛋白分析带来巨大的背景干扰以及使分离得到的蛋白结构可能发生改变,无法得到天然状态下的蛋白,给后续的确定工作带来障碍(sato,s.;etal.biochemicaltargetisolationfornovices:affinity-basedstrategies.chem.biol.,2010,17,616-623.)。
硼酸基团中的硼原子具有空的2p轨道,能够与亲核的杂原子(n、o、s等)形成可逆的共价结合。硼酸基团还能与具有1,2-二醇类结构的糖类化合物作用,形成ph相关型的硼酸酯类化合物,被应用于含有特定糖链结构的糖的识别及糖蛋白的研究中(pal,a.;etal.design,synthesis,andscreeningofalibraryofpeptidylbis(boroxoles)asoligosaccharidereceptorsinwater:identificationofareceptorforthetumormarkertf-antigendisaccharide.angew.chem.int.ed.,2010,49,1492-1495.)。研究发现,含有硼酸基团的抗肿瘤药bortezomib中的硼酸基团就能与靶标蛋白中的氨基酸残基形成相互作用,提高了药物的活性(touchet,s.;etal.aminoboronicacidsandestersfromsyntheticchallengestothediscoveryofuniqueclassesofenzymeinhibitors.chem.soc.rev.,2011,40,3895-3914.)。
kotoku等人设计合成含硼酸基团的探针分子(kotoku,n.etal..probemoleculeequippedwithboronicacidmoietyasareversiblecross-linkinggroupimprovesitsbindingaffinity.bioorg.med.chem.lett,2010,20,4152-4155.),证实硼酸基团可以提高探针分子与靶标蛋白的结合作用,但是由于活性小分子与靶标蛋白之间的结合存在不可逆性,捕捉得到的靶标蛋白不能在缓和的条件下,解离出来,得到天然构型的靶标蛋白,影响后续的靶蛋白的结构分析确定。因此,我们考虑设计一类新型可切断型的生物亲和探针分子。既能提高活性小分子与靶标蛋白间的亲和作用,又能使捕捉得到的靶标蛋白在缓和的条件下,解离出来,得到天然构型的靶标蛋白,有利于后续的靶蛋白的结构分析确定。
技术实现要素:
本发明通过合成一类生物亲和探针分子,能提高活性小分子与靶标蛋白间的亲和作用,又能使捕捉得到的靶标蛋白在缓和的条件下,解离出来,得到天然构型的靶标蛋白,可用于药用活性小分子靶标的发现及作用机制研究。本发明合成的化合物结构单一,合成方法简便,条件易于控制。
本发明的技术方案:
一类生物亲和探针分子,其为可逆式生物亲和探针分子化合物,具有如下通式i:
其中:
r1选自
r2选自需要进行靶标蛋白研究的具有药用活性的先导化合物;
n1为0~9中任一整数;n2为1~11中任一整数;
一种生物亲和探针分子化合物的制备方法,步骤如下:
(1)以化合物1为原料,溶解于thf,浓度为0.01~0.2m;n2保护,在0~5℃,依次加入化合物1a(氨基、叠氮基聚乙二醇化合物)、edci、hobt和三乙胺,化合物1:化合物1a:edci:hobt:三乙胺的摩尔比为1:2~4:2~4:2~4:2~4;在室温下反应2~20小时,得到化合物2;将化合物2溶解于二氯甲烷,浓度为0.01~0.2m,加入tfa,tfa:ch2cl2的体积比为0.1~0.5:1,在0~5℃反应10小时,得到化合物3;将化合物3溶解于thf,浓度为0.01~0.2m;依次加入频哪醇保护的硼酸的羧酸化合物、edci和hobt,化合物3:硼酸的羧酸化合物:edci:hobt的摩尔比为1:2~4:2~4:2~4:2~4;室温反应1~24小时,得到化合物4;将化合物4溶解于甲醇与盐酸水溶液的混合溶液,盐酸水溶液的ph为2~6,甲醇与盐酸水溶液体积比为1~4:1,化合物4的浓度为0.01~0.2m,再加入苯硼酸,化合物4:苯硼酸的摩尔比为1:1~6,室温反应1~24小时,得到硼酸基团脱保护的化合物5;将化合物5溶解于叔丁醇水溶液,叔丁醇与水的体积比为1~4:1,化合物5的浓度为0.01~0.2m,加入化合物5a(生物素炔丙胺化合物)、cuso4和抗坏血酸钠,化合物5:化合物5a:cuso4:抗坏血酸钠的摩尔比为1:1~5:1%~10%:1%~10%,反应1~24小时,得到化合物6;将化合物6溶解于体积比为1~4:1的四氢呋喃与水的混合溶液,浓度为0.01~0.2m,加入三(2-羰基乙基)磷盐酸盐,化合物6:三(2-羰基乙基)磷盐酸盐的摩尔比为1:1~2,在室温下反应2~10h,再加入溴代马来酰亚胺衍生化的活性小分子化合物,化合物6:溴代马来酰亚胺衍生化的活性小分子化合物的摩尔比为1:2~5,得到化合物7;结构式中的氨基、叠氮基聚乙二醇化合物的n1个数为0~9,脂肪直链n2碳的个数1~11;反应式如下:
(2)以化合物1为原料,溶解于thf,浓度为0.01~0.2m;n2保护,在0~5℃,依次加入化合物1a(氨基、叠氮基聚乙二醇化合物)、edci、hobt和三乙胺,化合物1:化合物1a:edci:hobt:三乙胺的摩尔比为1:2~4:2~4:2~4:2~4;在室温下反应2~20小时,得到化合物2;将化合物2溶解于二氯甲烷,浓度为0.01~0.2m,加入tfa,tfa:ch2cl2的体积比为0.1~0.5:1,在0~5℃反应10小时,得到化合物3;将化合物3溶解于thf,浓度为0.01~0.2m,依次加入含光反应性基团的的羧酸化合物、edci和hobt,化合物3:羧酸化合物:edci:hobt的摩尔比为1:2~4:2~4:2~4:2~4;室温反应1~24小时,得到化合物4;将化合物4溶解于叔丁醇水溶液,叔丁醇与水的体积比伟1~4:1,化合物4的浓度为0.01~0.2m,加入化合物5a(生物素炔丙胺化合物)、cuso4和抗坏血酸钠,化合物4:化合物5a:cuso4:抗坏血酸钠的摩尔比为1:1~5:1%~10%:1%~10%,反应1~24小时,得到化合物5;将化合物5溶解于体积比为1~4:1的四氢呋喃与水的混合溶液,化合物5的浓度为0.01~0.2m,加入三(2-羰基乙基)磷盐酸盐,化合物5:三(2-羰基乙基)磷盐酸盐的摩尔比为1:1~2,在室温下反应2~10h,再加入溴代马来酰亚胺衍生化的活性小分子化合物,化合物5:溴代马来酰亚胺衍生化的活性小分子化合物的摩尔比为1:2~5,得到化合物6;结构式中的氨基、叠氮基聚乙二醇化合物的n1个数为0~9,脂肪直链n2碳的个数1~11。反应式如下:
上述制备的可切断型的生物亲和探针分子用于药用活性先导化合物的靶标蛋白的发现及作用机制研究。
本发明所述的可切断型的生物亲和探针分子,能提高活性小分子与靶标蛋白间的亲和作用,又能使捕捉得到的靶标蛋白在缓和的条件下解离。可用于药用活性小分子靶标的发现及作用机制研究。本发明合成的化合物结构单一,合成方法简便,条件易于控制。
附图说明
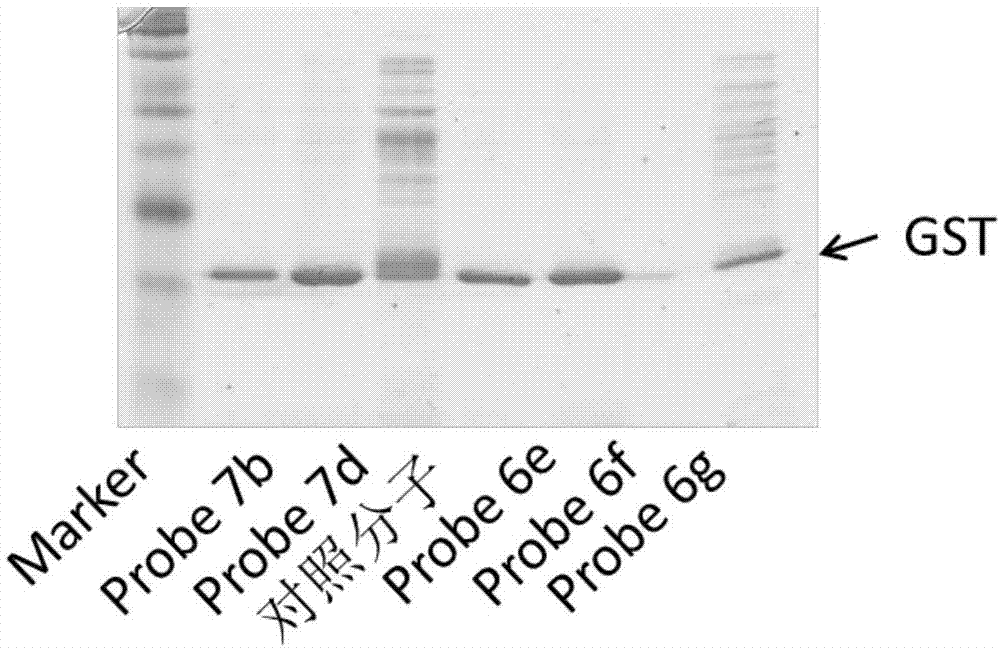
图1是生物亲和探针分子的性能评价图。
具体实施方式
下述非限制性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。
实施例1
化合物7b的合成
化合物1(750mg,3.4mmol),溶解于四氢呋喃(15ml),冰水浴搅拌下,加入化合物1‐氨基‐11‐叠氮基‐3,6,9‐三氧杂十一烷(800mg),edci(840mg),hobt(660mg),恢复室温反应12h。反应结束后,旋转蒸发仪浓缩,得到油状粗产品。粗产品柱层析,洗脱条件为(二氯甲烷/甲醇=30:1),得到化合物2(890mg),收率55%。化合物2:1hnmr(400mhz,cdcl3)d:7.67(brs,2h),5.58(d,j=9.2hz,2h),4.77(brs,2h),3.67–3.53(m,24h),3.47–3.41(m,4h),3.37(t,j=5.0hz,4h),2.95(m,4h),1.44(s,18h).esi‐msm/zforc32h61n10o12s2,[m+h]+calcd841.3912,found841.3936.
将化合物2(908mg),溶解于二氯甲烷(20ml),冰浴下缓慢滴加tfa(7ml),反应8h,旋转蒸发仪浓缩,得到粗产品,用25ml饱和碳酸氢钠溶解,用(二氯甲烷:甲醇=10:1)混合溶剂萃取三次,合并有机相,干燥,浓缩,得到淡黄色油状液体化合物3b(419mg),产率61%。化合物3:1hnmr(400mhz,cdcl3)d:7.62(brs,2h),5.57(d,j=9.2hz,2h),4.75(brs,2h),4.35(br,4h),3.69–3.55(m,24h),3.45–3.48(m,4h),3.28(t,j=5.0hz,4h),2.94(m,4h).esi‐msm/zforc22h45n10o8s2,[m+h]+calcd641.2863,found641.2841.
将化合物3(410mg),溶解于四氢呋喃(10ml),加入对羧基苯硼酸频哪醇酯(310mg),hobt(215mg),edci(301mg),室温反应10h,反应结束后,旋转蒸发仪浓缩,得到粗产品,粗产品柱层析,洗脱条件为(二氯甲烷/甲醇=30:1),得到化合物4b(546mg),产率78%。1hnmr(500mhz,cdcl3)d:7.88(4h,d,j=8.0hz),7.77(4h,d,j=8.0hz),5.72(br,2h),5.65(m,2h),3.56~3.43(m,28h),3.24(t,j=5.0hz,4h),3.07(m,4h),1.44(24h,s).esi‐msm/zforc56h83b2n10o14s2,[m+h]+calcd1205.5718,found1205.5688.
将化合物4b(250mg),溶解于甲醇与盐酸水溶液的混合溶液(10ml,盐酸水溶液的ph为3,甲醇与盐酸水溶液体积比为1:1),加入苯硼酸(101mg)室温过夜反应,加入饱和碳酸钾水溶液,调节ph约为9,二氯甲烷萃取三次,有机相干燥,浓缩,浓缩液柱层析,洗脱条件为(二氯甲烷/甲醇=30:1),得到化合物5b(160mg),产率82%。1hnmr(500mhz,cdcl3)d:7.88(4h,d,j=8.0hz),7.77(4h,d,j=8.0hz),5.72(br,2h),5.65(m,2h),3.56~3.43(m,28h),3.24(t,j=5.0hz,4h),3.07(m,4h).esi‐msm/zforc36h55b2n10o14s2,[m+h]+calcd937.3527,found937.3476.
将化合物5b(100mg)溶解于叔丁醇、水混合溶液(3ml,t‐buoh/h2o=1:1),加入生物素炔丙胺(217mg),cuso4水溶液(0.04m,30μl),氮气保护下,再加入抗坏血酸钠水溶液(10mm,0.6ml),氮气置换3次,30℃,600w微波反应30min,反应结束。反应液浓缩,硅胶柱层析,洗脱条件为(二氯甲烷/甲醇=15:1),得到化合物6b(130mg),收率81%。1hnmr(400mhz,cdcl3)δ7.89(4h,d,j=8.0hz),7.86~7.84(m,1h),7.81(s,2h),7.77(4h,d,j=8.0hz),7.28(d,j=5.6hz,2h),7.07(s,2h),6.39(s,2h),5.71(br,2h),5.66(m,2h),4.56~4.39(m,12h),4.35~4.29(m,2h),3.89(t,j=4.9hz,4h),3.66~3.50(m,22h),3.46~3.34(m,4h),3.15(d,j=4.6hz,2h),2.96(dd,j=13.2,6.0hz,2h),2.91(d,j=4.7hz,2h),2.76(d,j=12.8hz,2h),2.56(t,j=7.2hz,4h),2.23(t,j=7.2hz,4h).esi‐msm/zforc62h93b2n16o18s4,[m+h]+calcd1499.5923,found1499.5966.
将化合物6b(32mg)溶解于thf/h2o溶液(4:1,0.5ml),加入三(2‐羧乙基)膦盐酸盐(6.5mg),室温下搅拌2小时,待化合物6b反应完全后,加入谷胱甘肽溴化马来酰乙二胺化合物(30.3mg),室温反应10小时,反应结束后,浓缩,硅胶柱层析,洗脱条件为(二氯甲烷/甲醇=9:1),得到化合物7b(21mg),收率78%。1h‐nmr(400mhz,d2o)δ:7.88(2h,d,j=8.0hz),7.86(1h,s),7.77(2h,d,j=8.0hz),,7.07(1h,s),7.04(1h,s),4.76‐4.73(1h,m),4.57‐4.549(1h,m),4.51~4.49(3h,m),4.34(2h,s),4.26(1h,m),3.97(1h,m),3.95~3.79(2h,s),3.68‐3.60(6h,m),3.68‐3.47(12h,m),3.41‐3.26(3h,m),3.25‐3.05(6h,m),2.88(1h,dd,j=13.5,4.5hz),2.63(1h,d,j=12.8hz),2.58‐2.36(4h,m),2.20(2h,t,j=7.3hz),2.09(2h,m),1.56‐1.44(4h,m),1.18(3h,br).esi‐msm/zforc51h69bn13o19s3,[m+h]+calcd1274.4088,found1274.4132.
实施例2
化合物6e的合成
化合物1(750mg,3.4mmol),溶解于四氢呋喃(15ml),冰水浴搅拌下,加入化合物1‐氨基‐11‐叠氮基‐3,6,9‐三氧杂十一烷(800mg),edci(840mg),hobt(660mg),恢复室温反应12h。反应结束后,旋转蒸发仪浓缩,得到油状粗产品。粗产品柱层析,洗脱条件为(二氯甲烷/甲醇=30:1),得到化合物2(890mg),收率55%。化合物2:1hnmr(400mhz,cdcl3)d:7.67(brs,2h),5.58(d,j=9.2hz,2h),4.77(brs,2h),3.67–3.53(m,24h),3.47–3.41(m,4h),3.37(t,j=5.0hz,4h),2.95(m,4h),1.44(s,18h).esi‐msm/zforc32h61n10o12s2,[m+h]+calcd841.3912,found841.3936.
将化合物2(908mg),溶解于二氯甲烷(20ml),冰浴下缓慢滴加tfa(7ml),反应8h,旋转蒸发仪浓缩,得到粗产品,用25ml饱和碳酸氢钠溶解,用(二氯甲烷:甲醇=10:1)混合溶剂萃取三次,合并有机相,干燥,浓缩,得到淡黄色油状液体化合物3b(419mg),产率61%。化合物3:1hnmr(400mhz,cdcl3)d:7.62(brs,2h),5.57(d,j=9.2hz,2h),4.75(brs,2h),4.35(br,4h),3.69–3.55(m,24h),3.45–3.48(m,4h),3.28(t,j=5.0hz,4h),2.94(m,4h).esi‐msm/zforc22h45n10o8s2,[m+h]+calcd641.2863,found641.2841.
将化合物3(410mg),溶解于四氢呋喃(10ml),加入1‐羟基‐1,3‐二氢苯并[c][1,2]噁硼烷e‐6‐羧酸(285mg),hobt(215mg),edci(301mg),室温反应10h,反应结束后,旋转蒸发仪浓缩,得到粗产品,粗产品柱层析,洗脱条件为(二氯甲烷/甲醇=30:1),得到化合物4e(509mg),产率83%。1hnmr(500mhz,cdcl3)δ:8.02(2h,s),7.80(2h,d,j=8.0hz),7.42(2h,d,j=8.0hz),4.92(4h,s),5.72(br,2h),5.65(m,2h),3.56~3.43(m,28h),3.24(t,j=5.0hz,4h),3.07(m,4h),1.44(24h,s).esi‐msm/zforc38h55b2n10o14s2,[m+h]+calcd961.3527,found961.3575.
将化合物4e(100mg)溶解于叔丁醇、水混合溶液(3ml,t‐buoh/h2o=1:1),加入生物素炔丙胺(225mg),cuso4水溶液(0.04m,35μl),氮气保护下,再加入抗坏血酸钠水溶液(10mm,0.65ml),氮气置换3次,30℃,600w微波反应30min,反应结束。反应液浓缩,硅胶柱层析,洗脱条件为(二氯甲烷/甲醇=15:1),得到化合物5e(119mg),收率75%。1hnmr(400mhz,cdcl3)δ:8.02(2h,s),7.86~7.84(m,1h),7.81(s,2h),7.80(2h,d,j=8.0hz),7.42(2h,d,j=8.0hz),7.28(d,j=5.6hz,2h),7.07(s,2h),6.39(s,2h),5.71(br,2h),5.66(m,2h),4.92(4h,s),4.56~4.39(m,12h),4.35~4.29(m,2h),3.89(t,j=4.9hz,4h),3.66~3.50(m,22h),3.46~3.34(m,4h),3.15(d,j=4.6hz,2h),2.96(dd,j=13.2,6.0hz,2h),2.91(d,j=4.7hz,2h),2.76(d,j=12.8hz,2h),2.56(t,j=7.2hz,4h),2.23(t,j=7.2hz,4h).esi‐msm/zforc64h93b2n16o18s4,[m+h]+calcd1523.5923,found1523.5886.
将化合物5e(34mg)溶解于thf/h2o溶液(4:1,0.5ml),加入三(2‐羧乙基)膦盐酸盐(6.6mg),室温下搅拌2小时,待化合物6b反应完全后,加入谷胱甘肽溴化马来酰乙二胺化合物(30mg),室温反应10小时,反应结束后,浓缩,硅胶柱层析,洗脱条件为(二氯甲烷/甲醇=9:1),得到化合物6e(20mg),收率70%。1h‐nmr(400mhz,d2o)δ:8.06(1h,s),7.88(2h,d,j=8.0hz),7.86(1h,s),7.77(2h,d,j=8.0hz),7.07(1h,s),7.04(1h,s),4.92(2h,s),4.76‐4.73(1h,m),4.57‐4.549(1h,m),4.51~4.49(3h,m),4.34(2h,s),4.26(1h,m),3.97(1h,m),3.95~3.79(2h,s),3.68‐3.60(6h,m),3.68‐3.47(12h,m),3.41‐3.26(3h,m),3.25‐3.05(6h,m),2.88(1h,dd,j=13.5,4.5hz),2.63(1h,d,j=12.8hz),2.58‐2.36(4h,m),2.20(2h,t,j=7.3hz),2.09(2h,m),1.56‐1.44(4h,m),1.18(3h,br).esi‐msm/zforc52h69bn13o19s3,[m+h]+calcd1286.4088,found1286.4113.
应用例:
亲和探针靶蛋白吸附活性评价
将化合物(25μm)与过表达谷胱甘肽转移酶的大肠杆菌裂解液,加入磷酸缓冲溶液中(ph7.4,10mm),4℃,培养16小时,按探针分子比例,加入1.2倍量的链霉亲和素的琼脂糖珠,4℃,旋转混合2h,离心分离(1000rpm,4℃,3min),得到的琼脂糖珠加入磷酸缓冲液洗2次,再加入200μl蛋白洗脱液(20mmtris‐hclbuffer,100mmnacl,10mm2‐巯基乙醇),4℃,静置培养4小时,离心分离,取上清液进行进行蛋白质电泳实验。
如图1所示,所有的含有硼酸基团侧链的探针分子7b、7d,6e,6g在一定浓度的2‐巯基乙醇切断剂存在下,均可得到目标蛋白,而含有光亲和基团侧链化合物6g,只看到极少量的gst蛋白。推测是由于光亲和基团与蛋白表面的氨基酸残基发生不可逆的共价作用,即使加入切断剂,捕捉的靶标蛋白仍然不能与探针分子解离导致的,二硼酸基团的作用是可逆的,捕捉到的蛋白,可在切断剂存在下,解离出来。另外探针分子7d,6e,6g捕捉的蛋白要多于探针分子7b,说明硼酸基团数目及种类对探针分子与靶标蛋白的结合力均有影响。
综上所述,本发明所述的设计的一类新型可切断型的生物亲和探针分子。既能提高活性小分子与靶标蛋白间的亲和作用,又能使捕捉得到的靶标蛋白在缓和的条件下,解离出来,得到天然构型的靶标蛋白,有利于后续的靶蛋白的结构分析确定。本发明可用于具有药理活性的化合物的靶标蛋白的分离和发现。对研究活性化合物在生命体内的作用机制及新药开发具有重要的意义。本发明制备工艺简单,反应条件易于控制。
- 还没有人留言评论。精彩留言会获得点赞!