一种高通量检测植物circRNA等位位点差异表达的方法与流程
本发明属于基因表达检测技术领域,尤其涉及一种高通量检测植物circrna等位位点差异表达的方法。
背景技术:
等位基因(allele又作allelomorph)一般指位于一对同源染色体的相同位置上控制着相对性状的一对基因。
等位基因的不平衡表达(allelicexpressionimbalance,aei)为同一个细胞内,每个基因通常有2个拷贝,由于顺式作用使基因的2个拷贝的表达比例偏离了1:l。等位基因的不平衡表达现象普遍存在,除了遗传印迹基因的绝对不平衡表达外,有相当数量的基因在部分个体、同一个体不同时空上存在aei。并与基因组一些特定区域的多态位点相关。
目前,常用的等位基因不平衡表达检测主要集中于编码蛋白基因,而针对在转录组数据中广泛存在的circrna的等位表达情况尚未有高通量精准解析的方法。
技术实现要素:
有鉴于此,本发明的目的在于提供一种高通量检测植物circrna等位位点差异表达的方法。
为了实现上述发明目的,本发明提供了以下技术方案:
一种高通量检测植物circrna等位位点差异表达的方法,包括以下步骤:
1)提取植物样品总rna,利用所述总rna构建链特异性文库;
2)用illuminahiseq对步骤1)中所述链特异性文库进行双端测序,获得原始测序数据;
3)从步骤2)中获得的原始测序数据中筛选circrnas数据;
4)提取步骤3)中获得的circrnas数据中circrnas成环处的反向剪接reads;
5)对所述反向剪接reads进行单核苷酸变异检测获得所述反向剪接reads中的snp位点;
6)统计步骤4)中所述反向剪接reads中比对到步骤5)中所述snp位点的不同基因型的reads数,以比对到不同基因型的reads数的比例为不同基因型的表达量比例。
优选的,步骤3)中所述筛选circrnas数据包括以下步骤:
3.1)将所述原始测序数据据根据参考基因组进行转录本拼接;
3.2)将原始测序数据中未比对到参考基因组上的reads中的每一条read的两端提取18~22nt,组成一对anchor序列,所述anchor序列包括5’端序列和3’端序列;
3.3)将所述anchor序列与参考基因组序列进行重新比对,所述anchor序列的5'端序列比对到参考序列的3’端,所述anchor序列的3'端序列比对到所述参考序列中anchor序列的5'端序列的匹配位点的上游,并且在所述参考序列中在anchor序列的5’端序列匹配位点和anchor序列的3’端序列的匹配位点之间存在剪接位点gt-ag,则将此read作为circrna数据。
优选的,所述筛选circrnas数据通过find_circ软件和ciriexplorer软件实现。
优选的,利用find_circ软件和ciriexplorer软件分别筛选circrnas获得find_circ软件筛选出的circrnas候选数据和ciriexplorer软件筛选出的circrnas候选数据,取所述find_circ软件筛选出的circrnas候选数据和ciriexplorer软件筛选出的circrnas候选数据的交集作为circrnas数据。
优选的,步骤4)中提取获得的circrnas数据中circrnas成环处的反向剪接reads采用find_circ软件中samtoolsview–r指令实现。
优选的,步骤5)中所述单核苷酸变异检测采用gatk软件中的snpcalling进行。
优选的,步骤1)中所述植物样品总rna提取后、构建链特异性文库前,还包括依次进行的去除rrna步骤和线性rna消化步骤。
优选的,所述线性rna消化的反应体系为50μl,包括如下组分:rna,5μg;10×reactionbuffer,5μl;rnaser,20u;余量的rnase-freewater。
优选的,所述线性rna消化的温度为36~38℃,所述线性rna消化的时间为1~2h。
优选的,所述植物为林木。
本发明的有益效果:本发明所述方法可以针对转录组数据中广泛存在的circrna进行等位位点差异表达的高通量、准确的解析,为转录组数据的系统解析提供一个崭新的研究策略。
附图说明
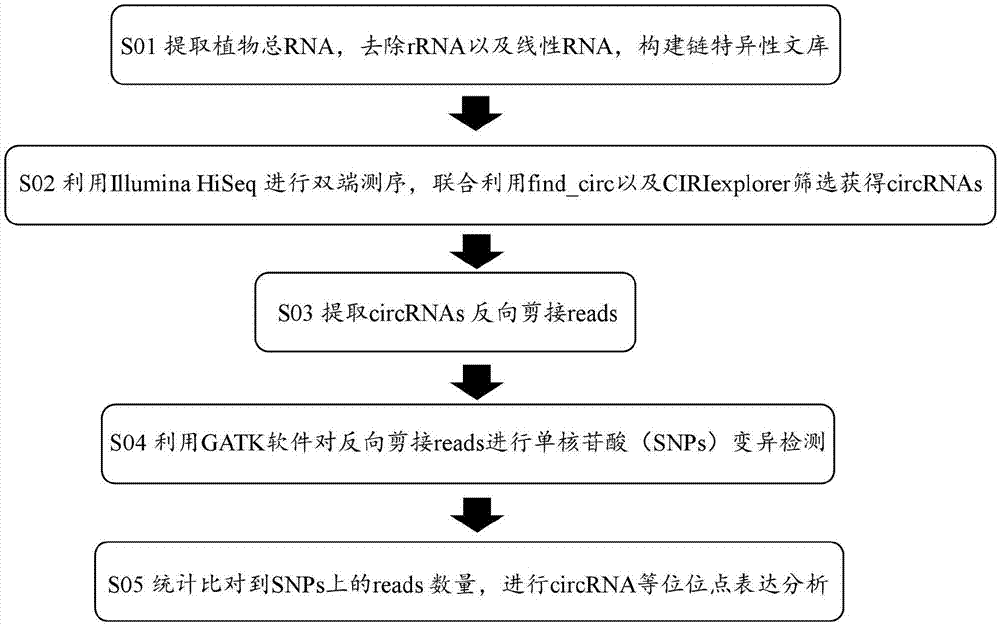
图1为植物circrna等位位点差异表达分析流程图。
具体实施方式
本发明提供了一种高通量检测植物circrna等位位点差异表达的方法,包括以下步骤:
1)提取植物样品总rna,利用所述总rna构建链特异性文库;
2)用illuminahiseq对步骤1)中所述链特异性文库进行双端测序,获得原始测序数据;
3)从步骤2)中获得的原始测序数据中筛选circrnas数据;
4)提取步骤3)中获得的circrnas数据中circrnas成环处的反向剪接reads;
5)对所述反向剪接reads进行单核苷酸变异检测获得所述反向剪接reads中的snp位点;
6)统计步骤4)中所述反向剪接reads中比对到步骤5)中所述snp位点的不同基因型的reads数,以比对到不同基因型的reads数的比例为不同基因型的表达量比例。
本发明提取植物样品总rna,利用所述总rna构建链特异性文库。本发明中对所述植物样品的种类没有特殊要求,常规植物均可,优选为林木,在本发明的具体实施过程中选用林木中的杨树。本发明对于所述植物样品优选为叶片组织。本发明对所述植物样品总rna的提取方法没有特殊限定,采用本领域常规的总rna提取方法即可,在本发明具体实施过程中,所述总rna的提取采用rna提取试剂盒(magjetplantrnapurificationkit,no.k2772)进行。
本发明在所述总rna提取后、构建链特异性文库前,优选的还包括依次进行的去除rrna步骤和线性rna消化步骤;所述去除rrna步骤优选的采用ribo-zerotmrrnaremovalkits(plant)试剂盒(no.mrzpl116)进行。本发明中,所述去除rrna的方法优选为:将30~50μl总rna与50~70μl磁珠混合,涡旋8~12s,室温静置4~6min,49~51℃孵育4~6min,置于磁力架上待上清液清澈,收集上清液;更优选为将40μl总rna与60μl磁珠混合,涡旋10s,室温静置5min,50℃孵育5min,置于磁力架上待上清液清澈2min,收集上清液。
本发明在所述去除rrna步骤后得到poly(a)-rna样品即线性rna,优选的对得到线性rna进行消化,所述消化优选的利用rnaserd进行;所述线性rna消化的反应体系优选为50μl,包括如下组分:rna,5μg;10×reactionbuffer,5μl;rnaser,20u;余量的rnase-freewater。所述线性rna消化的温度优选为36~38℃,更优选为37℃,所述线性rna消化的时间优选为1~2h,更优选为1.5h。本发明在所述消化后,利用消化后的rna构建链特异性文库,本发明中,所述构建链特异性文库优选的采用smart试剂盒(smartcdnalibraryconstructionkit,no.634901)。
本发明在获得所述连特异性文库后,用illuminahiseq对所述链特异性文库进行双端测序,获得原始测序数据。本发明中所述测序的读长优选为150nt;所述测序的数据量优选的大于12g;本发明中所述测序委托诺禾致源公司进行。
本发明在获得所述原始测序数据后,从获得的原始测序数据中筛选circrnas数据。在本发明具体实施过程中,首先去除原始测序数据中的接头以及冗余序列。本发明中,所述筛选circrnas数据包括以下步骤:
3.1)将所述原始测序数据进行转录本拼接;
3.2)将原始测序数据中未比对到参考基因组上的reads中的每一条read的两端提取18~22nt,组成一对anchor序列,所述anchor序列包括5’端序列和3’端序列;
3.3)将所述anchor序列与参考基因组序列进行重新比对,所述anchor序列的5'端序列比对到参考序列的3’端,所述anchor序列的3'端序列比对到所述参考序列中anchor序列的5'端序列的匹配位点的上游,并且在所述参考序列中在anchor序列的5’端序列匹配位点和anchor序列的3’端序列的匹配位点之间存在剪接位点gt-ag,则将此read作为circrna数据。
本发明中,优选的利用cufflinks软件默认参数进行转录本拼接;所述步骤3.2)和步骤3.3)优选的通过find_circ软件和ciriexplorer软件实现。更优选的,利用find_circ软件和ciriexplorer软件分别筛选circrnas获得find_circ软件筛选出的circrnas候选数据和ciriexplorer软件筛选出的circrnas候选数据,取所述find_circ软件筛选出的circrnas候选数据和ciriexplorer软件筛选出的circrnas候选数据的交集作为circrnas数据。
在本发明具体实施过程中,所述find_circ软件和ciriexplorer软件筛选circrnas的筛选参数包括-q5,-a20,-m2,-d2,--noncanonical。上述参数的筛选标准有选为:①-q5:anchor序列比对最小支持数5②-a20:anchor序列为20bp;③-m2:分支点不能在锚定序列(anchor)中2个核酸范围以内的其他地方出现;④-d2:序列比对只支持2个错配;⑤--noncanonical:gu/ag在剪切位点的两侧出现,并可以检测到明确的分支点(breakpoint)。
由于find_circ软件和ciriexplorer软件在筛选circrnas的过程中会产生假阳性数据,取所述find_circ软件筛选出的circrnas候选数据和ciriexplorer软件筛选出的circrnas候选数据的交集能够很大程度的降低假阳性,保证筛选到的circrnas数据的真实性和准确性。
本发明在获得所述circrnas数据后,提取所述circrnas数据中circrnas成环处的反向剪接reads;本发明中所述提取获得的circrnas数据中circrnas成环处的反向剪接reads优选的采用find_circ软件中samtoolsview–r指令实现。
本发明在获得所述反向剪接reads后,对所述反向剪接reads进行单核苷酸变异检测获得所述反向剪接reads中的snp位点;所述单核苷酸变异检测优选的采用gatk软件中的snpcalling进行。
本发明在获得所述反向剪接reads中的snp位点后,统计所述反向剪接reads中比对到所述snp位点的不同基因型的reads数,以比对到不同基因型的reads数的比例为不同基因型的表达量比例。
本发明所述方法通过上述步骤能够实现circrnas等位位点差异表达的高通量、高准确度的分析,为对后续circrnas的等位表达模式解析提供了技术支持,为全面破译基因植物基因组等位表达调控作用及基因组印记遗传效应奠定了基础,在植物复杂性状遗传效应解析以及分子设计育种等方面均具有较大的应用价值。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
采取毛白杨新鲜叶片,利用rna提取试剂盒(magjetplantrnapurificationkit,no.k2772)进行总rna提取,利用ribo-zerotmrrnaremovalkits(plant)试剂盒(no.mrzpl116)去除rrna,得到poly(a)-rna样品,利用rnaserd对线性rna进行消化(反应体系:rna,5μg;10xreactionbuffer,5μl;rnaser,20u;rnase-freewater,补充至50μl),获得poly(a)-/ribo-rna样品,利用smart试剂盒(smartcdnalibraryconstructionkit,no.634901)进行链特异性cdna文库构建;
利用illuminahiseqtm2500进行双端测序,测序数据量为12g。去除接头以及冗余序列,通过cufflinks软件默认参数拼接转录本。利用find_circ对没有比对到参考序列(参考序列为毛果杨v3.0版基因组序列https://phytozome.jgi.doe.gov/pz/portal.html)的序列,两端各提取20-nt作为一对anchor序列,将每一对anchor序列再次比对参考序列,如果anchor序列的5'端比对到参考序列(起始与终止位点分别记为a3,a4),同时该anchor序列的3'端比对到anchor序列5'端匹配位点的上游(起始与终止位点分别记为a1,a2),并且在参考序列的a2到a3之间存在剪接位点(gt-ag),则将此read作为候选circrna。筛选参数:-q5,-a20,-m2,-d2,--noncanonical。筛选标准:①-q5:anchor序列比对最小支持数5②-a20:anchor序列为20bp;③-m2:分支点不能在锚定序列(anchor)中2个核酸范围以内的其他地方出现;④-d2:序列比对只支持2个错配;⑤--noncanonical:gu/ag在剪切位点的两侧出现,并可以检测到明确的分支点(breakpoint)。同时,利用ciriexplorer软件的默认参数对circrna进行筛选。find_circ软件分析获得887个circrnas,利用ciriexplorer软件获得920个circrnas,根据circrnas反向剪接reads对两个预测结果取交集,共获得97个circrna(表1)。
表1毛白杨叶片候选circrna
根据find_circ分析结果,利用samtoolsview–r指令提取取circrnas成环处的反向剪接reads用于后续的核酸变异分析。
对提取的reads序列,利用gatk(version:4.0.1.0)软件进行snpcalling,步骤如下:首先利用软件中haplotypecaller工具进行2个样本的变异检测,--pair-hmm-gap-continuation-penalty参数设置为10,其余参数均为默认值,得到各个样本的变异信息,利用combinegvcfs工具将各样本的变异文件合并。最后利用genotypegvcfs工具进行各样本间的等位变异检测,生成一个vcf文件,vcf文件中会包含所有样本的变异位点和基因型信息(表2)。
利用反向剪接reads中的snps作为标记,统计比对snps上的反向剪接reads数量作为候选circrna等位位点的表达量(表2)。
表2毛白杨叶片候选circrna等位位点表达模式
结果显示在毛白杨叶片中仅有44.7%的circrna等位位点平衡表达,其余位点均为不平衡表达。
实施例2
采取小叶杨高温处理叶片用于总rna提取,利用rna提取试剂盒(magjetplantrnapurificationkit,no.k2772),利用ribo-zerotmrrnaremovalkits(plant)试剂盒(no.mrzpl116)去除rrna,随后利用磁珠法结合poly(a)的rna,得到poly(a)-rna样品,利用rnaserd对线性rna进行消化,(反应体系:rna,5μg;10xreactionbuffer,5μl;rnaser,20u;rnase-freewater,补充至50μl),获得poly(a)-/ribo-rna样品,利用smart试剂盒(smartcdnalibraryconstructionkit,no.634901)进行链特异性cdna文库构建;
利用illuminahiseqtm2500进行双端测序,测序数据量为12g。去除接头以及冗余序列,通过cufflinks软件拼接转录本。利用find_circ对没有比对到参考序列的reads的两端各提取20-nt的anchor序列,将每一对anchor序列再次比对参考序列,如果anchor序列的5'端比对到参考序列(起始与终止位点分别记为a3,a4),同时该anchor序列的3'端比对到此位点的上游(起始与终止位点分别记为a1,a2),并且在参考序列的a2到a3之间存在剪接位点(gt-ag),则将此read作为候选circrna。筛选参数:-h,-v,-s,-g,-n,-p,-q,-a,-m,-d,--noncanonical,--randomize,--allhits,--stranded,--strandpref,--halfunique。筛选参数包括-q5,-a20,-m2,-d2,--noncanonical。上述参数的筛选标准有选为:①-q5:anchor序列比对最小支持数5②-a20:anchor序列为20bp;③-m2:分支点不能在锚定序列(anchor)中2个核酸范围以内的其他地方出现;④-d2:序列比对只支持2个错配;⑤--noncanonical:gu/ag在剪切位点的两侧出现,并可以检测到明确的分支点(breakpoint)同时,利用ciriexplorer软件的默认参数对circrna进行筛选。find_circ软件分析获得804个circrnas,利用ciriexplorer软件获得670个circrnas,根据circrnas反向剪接reads对两个预测结果取交集,共获得121个circrna(表3)。
表3小叶杨高温响应circrna
根据find_circ分析结果,整理circrnas成环处的反向剪接reads数据标签文件,利用samtoolsview–r指令提取取circrnas成环处的反向剪接reads用于后续的核酸变异分析。
对提取的reads序列,利用gatk(version:4.0.1.0)软件进行snpcalling,步骤如下:首先利用软件中haplotypecaller工具进行2个样本的变异检测,--pair-hmm-gap-continuation-penalty参数设置为10,其余参数均为默认值,得到各个样本的变异信息,利用combinegvcfs工具将各样本的变异文件合并。最后利用genotypegvcfs工具进行各样本间的等位变异检测,生成一个vcf文件,vcf文件中会包含所有样本的变异位点和基因型信息(表2)。
利用反向剪接reads中的snps作为标记,统计比对snps上的反向剪接reads数量作为候选circrna等位位点的表达量(表4)。
表4小叶杨高温响应circrna等位位点表达模式
结果显示在小叶杨高温胁迫处理的叶片组织中仅有25.8%的circrna等位位点平衡表达,其余位点均为不平衡表达。
由以上实施例可知,本发明所提供的方法,采用链特异性文库rna测序,联合利用circrna分析软件及核酸变异分析软件,可以高通量精准解析植物circrna等位位点的表达模式,根据实施例中结果可知,所述方法可以实现植物circrna等位表达的高通量解析,为充分利用转录组测序结果,系统解析circrna等位表达模式提供了新的研究方案。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!