在哺乳动物干细胞中进行基因组编辑的方法与流程
本发明涉及一种在哺乳动物干细胞中进行基因组编辑的方法。
背景技术:
人胚胎干细胞(esc)因其具有无限的自我更新能力可为再生医学提供充足的细胞来源,但其衍生自胚泡,这存在伦理问题。患者特异性诱导的多能干细胞(ipsc)的发现解决了与同种异体细胞的移植有关的免疫原性问题和伦理问题。近来,采用表达重编程因子的非整合型载体从外周血等易于获得的细胞来源生成ipsc已经取得了很大进展。然而,为了实现ipsc在再生医学和疾病建模中的潜力,在治疗前通常需要对致病基因进行纠正或修饰。虽然这些方法是通用的,但是它们常常会导致整个供体质粒被整合,并且可能会诱发由错误的nhej引起的突变连接,从而限制了应用前景,因此,研究用于进行高效精确基因敲入的新方法是当前的一个重要课题。
几十年前即已实现了在小鼠esc内进行基因打靶,尽管效率极低。进一步的研究使人们认识到,早期的成就已经在无意间采用了dna断裂后细胞固有的修复机制。然而,天然发生的围绕靶向基因座的双链dna断裂(dsb)是极其罕见的,因此,即使使用长度为10千碱基对的同源臂,靶向效率也常常被限制在百万分之一的水平。为了增强基因打靶,人们在过去的二十年一直致力于通过可靶向内切酶在特定基因座创建dsb。虽然锌指核酸酶(zfn)或转录激活子样效应核酸酶(talen)等工程化核酸内切酶的发展已经引起了人们的兴趣,但其在设计或克隆方面的局限性使它们不可能用于常规实验室。最新一代rna引导的核酸内切酶或crispr-cas9因其载体设计简单、性能稳定而得到广泛应用。crispr-cas9是一种在细菌和古细菌中进化而来的适应性免疫系统,用以识别和破坏噬菌体或质粒等侵入剂。常用的cas9来自化脓性链球菌(sp)。
创建双链dna断裂(dsb)后,细胞机制主要通过两种途径来修复损伤:非同源末端连接(nhej)和同源定向修复(hdr)。在不存在模板的条件下,利用nhej途径,从而在dsb位点进入可变插入或缺失(indels),这可能会破坏基因的开放阅读框架并生成基因敲除(ko)表型。这种编辑方法效率较高,且已广泛用于动物建模、疾病建模和功能基因组研究中。在存在与同源臂(ha)侧翼连接的供体模板的条件下,hdr途径可以用于整合ha之间的序列以形成精确的dna缺失、取代或插入,从而修正患病基因或靶向的整合目的基因。不幸地是,hdr介导的基因敲入的效率通常较低。
人psc的编辑效率低下主要是由于实验操作后存活率低。与小鼠psc相反,将人psc分离为单细胞悬浮液通常会引起大量细胞死亡。通过防止失巢凋亡,即分离引起的细胞凋亡,rock抑制因子的使用可显著增加克隆效率。然而,这只能解决一个问题。为了精确地编辑psc,需要将crispr组分,即cas9和sgrna,以及dna供体模板一起输送至细胞中。最有效的载体输送方式为电穿孔或其改进的核转染,但这会引起细胞死亡。
在质粒载体核转染期间或之后,由于大量细胞死亡仍是ipsc基因组编辑的主要障碍,因而研究在多能干细胞中进行基因组编辑的新方法是当前主要的课题。
技术实现要素:
本发明提供了一种用于进行高效、精确基因编辑的方法。
在一实施方案中,本文公开了一种在哺乳动物干细胞中编辑目标基因组dna的方法。所述方法包括将细胞凋亡调控因子和基因组编辑组合物引入哺乳动物干细胞中,生成过表达细胞凋亡调控因子的经修饰的哺乳动物干细胞,其中所述细胞凋亡调控因子为bcl-xl,其中所述基因组编辑组合物为基因组核酸内切酶,所述基因组核酸内切酶在哺乳动物干细胞的基因组dna的所需目标序列内进行切割,并编辑目标基因组dna。
在一些实施方案中,所述bcl-xl包含具有与seqidno:1的氨基酸序列有至少70%氨基酸序列相同的氨基酸序列。
在一些实施方案中,将细胞凋亡调控因子和基因组编辑组合物引入哺乳动物干细胞中包括:将所述细胞凋亡调控因子引入所述哺乳动物干细胞中,生成所述过表达所述细胞凋亡调控因子的经修饰的哺乳动物干细胞;以及将所述基因组编辑组合物引入所述经修饰的哺乳动物干细胞中。
在一些实施方案中,所述基因组编辑组合物可包括rna-引导的核酸内切酶和引导rna。
在一些实施方案中,所述rna-引导的核酸内切酶可以为cas9。
在一些实施方案中,所述引导rna可包括成簇的规律间隔的短回文重复rna和tracrrna。
在一些实施方案中,所述基因组编辑组合物可包括包含供体序列的供体质粒,其中,通过同源定向修复在插入位点将所述供体序列插入基因组中。
在一些实施方案中,所述哺乳动物干细胞可包括胚胎干细胞或多能干细胞。
在一些实施方案中,所述哺乳动物干细胞可以为诱导的多能干细胞。
在一些实施方案中,所述方法还可包括将bcl抑制因子引入细胞中。
在一些实施方案中,所述bcl抑制因子可以为abt-263。
在一些实施方案中,所述bclxl被稳定表达。
在某些实施方案中,所述bclxl被瞬时过表达。
在一些实施方案中,所述bclxl过表达大约1小时至大约72小时。
在一些实施方案中,在背景上,所述bclxl被过表达至少5倍。
附图说明
图1示出了在人ipsc中稳定过表达bcl-cl的方案设计。
图2是在prdm14处进行hdr编辑的示意图。
图3示出了crispr质粒共转染后ipsc-lenti-bcl-xl系和ipsc-lenti-对照系的流式细胞术分析。
图4示出了在人ipsc中进行瞬时bcl-cl过表达的方案设计。
图5示出了瞬时bcl-xl过表达组和对照组的流式细胞术分析。
图6是在ctnnb1处进行hdr编辑的示意图。
图7是在oct4处进行hdr编辑的示意图。
图8示出了指示ipsc中ctnnb1和oct4处的ki效率的流式细胞术分析。
图9是在cd326处进行nhej介导的基因敲除的示意图。
图10是在cd9处进行nhej介导的基因敲除的示意图。
图11示出了指示ipsc中cd326和cd9处的ko效率的流式细胞术分析。
图12示出了用基因组编辑质粒在结合或不结合bcl-xl下进行电穿孔后相对细胞数的动态变化。
图13示出了结合bcl-xl、bcl2或mcl1的ipsc的流式细胞术分析。
图14示出了cas9和sgrna的表达水平。
图15示出了bax基因敲除对ipsc细胞存活率和编辑效率的影响。
图16示出了bbc3基因敲除对ipsc细胞存活率和编辑效率的影响。
图17示出了电穿孔后abt-263的不同基剂量和疗程对ipsc的细胞存活率的影响。
图18示出了电穿孔后abt-263的不同基剂量和疗程对ipsc的hdr效率的影响。
图19示出了abt-263对ipsc细胞存活率和编辑效率的影响。
图20是ipsc中prdm14和ctnnb1处的hdr介导的双重ki的示意图。
图21示出了指示ipsc中ctnnb1处的ki效率的流式细胞术分析。
图22是通过hdr在prdm14处和通过nhej在cd326处进行双重编辑的示意图。
图23示出了指示ipsc中cd326处的ko效率的流式细胞术分析。
图24是通过切片盒的等位基因hdr插入进行基因敲除的示意图。
图25示出了指示ipsc中cd326处的ko效率的流式细胞术分析。
图26示出了小鼠es细胞中瞬时bcl-xl、bcl2或mcl1过表达对hdr效率的影响。
图27示出了k562细胞中瞬时bcl-xl、bcl2或mcl1过表达对hdr效率的影响。
图28示出了小鼠193t细胞中瞬时bcl-xl、bcl2或mcl1过表达对hdr效率的影响。
图29示出了小鼠jurkat细胞中瞬时bcl-xl、bcl2或mcl1过表达对hdr效率的影响。
具体实施方式
本发明提供了一种新的基因组编辑方法,该方法的效率比采用crispr/cas9等rna引导的核酸内切酶的传统基因组编辑方法的效率显著更高。本申请的方法采用细胞凋亡调节因子bcl-xl。通过在ipsc转染期间过表达bcl-xl,可以在电穿孔后提高ipsc的存活率,并且还能提高hdrki效率和ko效率。改进的基因组编辑系统为从操纵人ipsc基因组到创建基因修饰的动物模型的应用提供有用的工具。
术语“核酸”、“多核酸”和“寡核苷酸”可交换使用,是指以线性或圆形构象和单链或双链形式存在的脱氧核糖核酸或核糖核苷酸聚合物。针对本发明的目的,这些术语不应视为是限制聚合物的长度。所述术语可包含已知的天然核苷酸类似物以及在碱基、糖和/或盐酸酯部分(例如,硫代磷酸酯骨架)被修饰的核苷酸。一般来说,具体的核苷酸类似物具有相同的碱基配对特异性;即,a的类似物将与t进行碱基配对。
术语“多肽”、“肽”和“蛋白质”可互换使用,是指具有氨基酸残基的聚合物。所述术语还可适用于氨基酸聚合物,其中,一个或多个氨基酸是相应的天然存在的氨基酸的化学类似物或经修饰的衍生物。
术语“序列”是指任意长度的核苷酸序列,其可以是dna或rna;可以是线性的、圆形的或分支的,并且可以是单链的或是双链的。
本文使用的术语“同源核酸”包含与已知的参考序列相同或相似的核酸序列。在一实施方案中,使用术语“同源核酸”来表征与已知参考序列有至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%甚或是100%相同的序列。
术语“同源定向修复(hdr)”是指特殊形式的dna修复,例如,它发生在修复细胞内双链断裂期间。该过程要求核苷酸序列具有同源性,使用“供体”分子来模板修复“目标”分子(即,经历双链断裂的分子),并且使遗传信息从供体转移至目标。如果供体核苷酸不同于目标分子并且供体核苷酸的部分或全部序列包含在目标dna中,则同源定向修复可导致目标分子的序列发生变化(例如,插入、缺失、突变)。
术语“非同源末端连接(nhej)”是指通过将断裂端直接连接到另一个断裂端来修复dna中的双链断裂,不需要同源模板(与同源定向修复不同,同源定向修复要求同源序列来指导修复)。nhej通常会导致双链断裂位点附近的核苷酸序列丢失(缺失)。
在本文中,术语“干细胞”用于指一种具有能自我更新并且生成分化的细胞型的能力的细胞(例如,植物干细胞、脊椎动物干细胞)。在细胞个体发生的背景下,形容词“分化的”或“分化”是一个相对的术语。“分化的细胞”是一种在发育过程中比与其比较的细胞进展地更远的细胞。
在本文中,术语“多能干细胞”或“psc”用于意指一种能够产生所有有机体细胞类型的干细胞。因此,psc可产生所有有机体生殖层(例如,脊椎动物的内胚层、中胚层和外胚层)的细胞。多能细胞能形成畸胎瘤并且有益于活的有机体内的内胚层、中胚层和外胚层组织。植物的多能干细胞能产生植物的所有细胞类型(例如,根、茎、叶子等的细胞)。由于术语psc是指多能干细胞,不管其来源如何,术语psc包括术语esc和ipsc,以及术语胚胎生殖干细胞(egsc),这些是psc的另一个例子。psc可以是已建立的细胞系的形式,它们可以直接从原始胚胎组织中获得,也可以从体细胞中获得。psc可以是本文所述方法的目标细胞。
术语“诱导的多能干细胞”或“ipsc”是指一种衍生自不是psc的细胞的psc(即,衍生自相对于psc而分化的细胞)。ipsc可衍生自多种不同的细胞类型,包括终末分化细胞。ipsc具有es细胞样形态,生长为扁平集落,核质比大,边界清晰,细胞核显著。
如本文使用的术语“供体序列”是指一种待插入宿主供体序列的染色体中的核酸。供体序列可以是任意长度,例如,长度为2至10,000个核苷酸(或其之间或以上的任意整数值)。
本文提及的“大约”值或参数包括(并且描述)针对该值或参数本身的变化。例如,对“大约x”的描述包括对“x”的描述。
如本文和所附权利要求中所使用的,除非上下文另有明确规定,否则单数形式的“一”、“或”和“所述”包括复数个指示对象。
本发明的组合物和方法可包括、由或基本上由本文所述的本发明的基本要素和限制以及本文所述的或用于营养或制药用途的任何附加或任选成分、组分或限制组成。
本发明提供了一种在哺乳动物干细胞中进行基因组编辑的方法。所述方法包括通过过表达细胞凋亡调节因子来修饰哺乳动物干细胞和将基因组编辑组合物引入哺乳动物干细胞中。基因组编辑包括nhej和hdr。编辑基因组的核酸内切酶可在目标基因组dna中产生单或双链断裂,该单或双链断裂被修复。通过nhej发生的修复有时是指“indel”(插入或缺失);通过hdr发生的dna修复有时被称为“基因修正”或“基因修饰”。在某些情况下,编辑目标基因组dna包括取代目标基因组dna中的一个或多个核苷酸,从而生成编辑的目标基因组dna。在某些情况下,编辑目标基因组dna包括从目标基因组dna中删除一个或多个核苷酸,从而生成编辑的目标基因组dna。在某些情况下,编辑目标基因组dna包括从目标基因组dna中插入一个或多个核苷酸,从而生成编辑的目标基因组dna。
在一些实施方案中,所述方法包括将细胞凋亡调节因子和基因组编辑组合物引入哺乳动物干细胞中,生成过表达细胞凋亡调节因子的经修饰的哺乳动物干细胞,其中所述细胞凋亡调节因子为bcl-xl,其中所述基因组编辑组合物为基因组编辑核酸内切酶,所述基因组核酸内切酶在哺乳动物干细胞的基因组dna的所需目标序列内进行切割,并编辑目标基因组dna。由bcl2样1(bcl1l1)基因编码的bcl-xl可保持线粒体外膜的完整性,从而防止细胞色素c、细胞凋亡活化因子等线粒体内容物的释放。
在一些实施方案中,bcl-xl包含与seqidno:1的氨基酸序列有至少70%、至少90%、至少95%、至少98%、至少99%或至少100%氨基酸序列相同的氨基酸序列。
在一些实施方案中,本文所述的哺乳动物干细胞可以是胚胎干细胞或多能干细胞。在一些实施方案中,哺乳动物干细胞可以是诱导的多能干细胞。在一些实施方案中,哺乳动物干细胞可以是人诱导的多能干细胞。
可采用已知的方法实现bcl-xl在哺乳动物干细胞中的过表达。在某些情况下,将bcl-xl以蛋白质的形式引入哺乳动物干细胞中。在某些情况下,将bcl-xl以信使rna(mrna)或cdna等核酸的形式引入哺乳动物干细胞中。核酸可以作为质粒或病毒载体等较大构建体的一部分输送,或者例如直接通过电穿孔、脂质囊泡、病毒转运蛋白和显微注射输送。在一些实施方案中,可通过包括转染、磷酸钙-dna共沉淀、deae-葡聚糖介导的转染、聚凝胺介导的转染、电穿孔、显微注射、转导、细胞融合、脂质体融合、脂质转染、原生质体融合、逆转录病毒感染、使用基因枪、使用dna载体转运子和生物弹(例如,粒子轰击)在内的各种本领域已知的手段将bcl-xl引入细胞中。在一些实施方案中,编码bcl-xl的核苷酸序列可操作地与启动子等转录控制元件连接,其中所述启动子在哺乳动物干细胞中是活跃的。在某些情况下,启动子为组成型启动子。在这种情况下,bclxl表达稳定。在某些情况下,启动子为诱导型启动子。在一些实施方案中,将编码bcl-xl的核苷酸序列引入哺乳动物干细胞中,然后再插入哺乳动物干细胞的基因组中。
在一些实施方案中,将基因组编辑组合物和bcl-xl同时引入细胞中。在一些实施方案中,可先将bcl-xl引入细胞中,然后再引入基因组编辑组合物。在一些实施方案中,将细胞凋亡调节因子(例如,bcl-xl)和基因组编辑组合物引入哺乳动物干细胞中包括以下步骤。首先,将细胞凋亡调节因子(例如,bcl-xl)引入哺乳动物干细胞中,生成过表达细胞凋亡调节因子的经修饰的哺乳动物干细胞。然后,将基因组编辑组合物引入所述经修饰的哺乳动物干细胞中。
在一些实施方案中,bcl-xl组成型地过表达在细胞中。在一些实施方案中,bcl-xl被瞬时过表达。例如,在某些情况下,bcl-xl过表达大约1小时至大约72小时、大约4小时至大约8小时、大约8小时至大约12小时、大约12小时至大约16小时、大约16小时至大约20小时、大约20小时至大约24小时、大约24小时至大约30小时、大约30小时至大约36小时、大约36小时至大约42小时或者大约42小时至大约48小时。在某些情况下,bcl-xl过表达大约12小时至大约48小时。在某些情况下,bcl-xl过表达大约12小时至大约24小时。
在一些实施方案中,将bcl-xl引入细胞中,以使细胞中bcl-xl的含量是对照(未经修饰)细胞中bcl-xl的背景含量的至少2倍,或高于2倍。例如,在某些情况下,将bcl-xl引入细胞中,以使得细胞中bcl-xl的含量比对照(未经修饰)细胞中bcl-xl的背景含量高2倍至10倍或5倍至10倍。
在哺乳动物干细胞中过表达bcl-xl可生成经修饰的哺乳动物干细胞。因此,可在bcl-xl被过表达期间将基因组编辑组合物引入哺乳动物干细胞中。
在一些实施方案中,当哺乳动物干细胞与基因组编辑组合物接触时,bcl-xl在哺乳动物干细胞中的过表达使哺乳动物干细胞的存活率比未过表达bcl-xl并与基因组编辑组合物接触的对照干细胞的存活率增加至少5倍、至少10倍、至少20倍或者超过20倍。例如,在某些情况下,当哺乳动物干细胞与基因组编辑组合物接触时,bcl-xl在哺乳动物干细胞中的过表达使哺乳动物干细胞的存活率比未过表达bcl-xl并与基因组编辑组合物接触的对照干细胞的存活率增加10倍至20倍。
在一些实施方案中,当哺乳动物干细胞与基因组编辑组合物接触时,bcl-xl在哺乳动物干细胞中的过表达使hdr介导的ki(基因敲入)效率比未过表达bcl-xl并与基因组编辑组合物接触的对照干细胞的存活率增加至少10倍、至少20倍、至少50倍、至少100倍或者高于100倍。例如,在某些情况下,当哺乳动物干细胞与基因组编辑组合物接触时,bcl-xl在哺乳动物干细胞中的过表达使hdr介导的ki(基因敲入)效率比未过表达bcl-xl并与基因组编辑组合物接触的对照干细胞的存活率增加20倍至100倍。
在一些实施方案中,当哺乳动物干细胞与基因组编辑组合物接触时,bcl-xl在哺乳动物干细胞中的过表达使nhej介导的ko(基因敲除)效率比未过表达bcl-xl并与基因组编辑组合物接触的对照干细胞的存活率增加至少1倍、至少2倍、至少5倍或者高于5倍。例如,在某些情况下,当哺乳动物干细胞与基因组编辑组合物接触时,bcl-xl在哺乳动物干细胞中的过表达使nhej介导的ko(基因敲除)效率比未过表达bcl-xl并与基因组编辑组合物接触的对照干细胞的存活率增加1倍至5倍。因此,当哺乳动物干细胞过表达bcl-xl时,哺乳动物干细胞的基因组编辑效率增加。
在一些实施方案中,基因组编辑组合物包括rna引导的核酸内切酶和引导rna。
在一些实施方案中,将rna引导的核酸内切酶以蛋白质的形式或者以信使rna(mrna)或cdna等编辑rna引导的核酸内切酶的核酸的形式引入真核细胞中。核酸可作为质粒或病毒载体等较大构建体的一部分输送,或者例如直接通过电穿孔、脂质囊泡、病毒转运蛋白和显微注射输送。可通过包括转染、磷酸钙-dna共沉淀、deae-葡聚糖介导的转染、聚凝胺介导的转染、电穿孔、显微注射、转导、细胞融合、脂质体融合、脂质转染、原生质体融合、逆转录病毒感染、使用基因枪、使用dna载体转运子和生物弹(例如,粒子轰击)在内的各种本领域已知的手段将rna介导的核酸内切酶引入细胞中。
在一些实施方案中,可通过转染(包括,例如,通过电穿孔的转染)将编辑rna引导的核酸内切酶的核酸引入细胞中。在一些实施方案中,可通过注射将编辑rna引导的核酸内切酶的核酸引入细胞中。
在一些实施方案中,例如,可引入作为rna或作为质粒或其他编辑引导rna的核酸载体的引导rna(grna)。rna引导的核酸内切酶与grna和与之连接的目标dna结合并在设计的特殊位点切割染色体。例如,可通过包括转染、磷酸钙-dna共沉淀、deae-葡聚糖介导的转染、聚凝胺介导的转染、电穿孔、显微注射、转导、细胞融合、脂质体融合、脂质转染、原生质体融合、逆转录病毒感染、使用基因枪、使用dna载体转运子和生物弹(例如,粒子轰击)在内的各种本领域已知的手段将引导rna(grna)引入细胞中。
在一些实施方案中,rna引导的核酸内切酶位序列特异性核酸酶。本文所用的术语“序列特异性核酸酶”是指一种识别并与特殊核酸序列处的多核苷酸结合以及催化该多核苷酸内的双链断裂的蛋白质。在某些实施方案中,rna引导的核酸内切酶仅切割染色体一次,即,在本文所述的方法中,在设计的特殊位点引入单一的双链断裂。
可与本文所述方法和组合物一起使用的rna引导的核酸内切酶系统的示例包括cas/crispr系统。cas/crispr(成簇的规律间隔的短回文重复)系统采用目标dna的rna引导的dna结合与序列特异性切割。引导rna(grna)含有大约20-25(如20)个与位于基因组pam(原间隔基序)位点上游的目标基因组dna序列和恒定rna支架区互补的核苷酸。在某些实施方案中,目标序列与pam有关,pam是由crispr复合物识别的短序列。pam的精确序列和长度要求会依据所使用的crispr酶而不同,但pam通常为与前间隔序列(即,目标序列)相邻的2-5bp序列。pam序列的示例在本领域是公知的,本领域的技术人员将能进一步识别用于给定的crispr酶的pam序列。例如,具有pam序列ngg的化脓性链球菌(s.pyogenes)的cas9的目标位点可通过在输入序列和输入端的反向补体上搜索5'-nx-ngg-3'来识别。在某些实施方案中,本文所用的基因组pam位点为ngg、nng、nag、nggng或nnagaaw。在具体的实施方案中,使用s.pyogenescas9(spcas9),对应的pam为ngg。在某些方面,来自不同菌株的不同cas9酶采用不同的pam序列。cas(与crispr相关的)蛋白质与grna和与之连接的目标dna连接并在pam位点上游的限定位置引入双链断裂。在一方面,crispr/cas、cas/crispr或者crispr-cas系统(这些术语在整个应用中可互换使用)不要求生成定制的蛋白质来靶向特定序列,而是由短rna分子编程单个cas酶以识别特定的dna靶标,即,使用短rna分子将cas酶招募到特定的dna靶标中。
在一些实施方案中,rna引导的核酸内切酶为ii型cas蛋白质。在一些实施方案中,rna引导的核酸内切酶为cas9,其同系物或其经修饰的版本。在一些实施方案中,可以使用两个或两个以上cas蛋白质的组合。在一些实施方案中,crispr酶为cas9,也可以是来自s.pyogenes或肺炎链球菌s.pneumoniae的cas9。在一些实施方案中,在本文所述的方法中使用cas9。cas9含有两个与hnh和ruvc核酸内切酶同源的独立的核酸酶结构域,并且能在grna的引导下切割dna的双链,生成钝头双链断裂(dsb)。
在一些实施方案中,引导rna是包含5’区和3’区的rna,所述5’区含有来自crispr基因座的至少一个重复序列,所述3’区与染色体上的预定插入位点互补。在某些实施方案中,5’区含有与染色体上的预定插入位点互补的序列,3’区含有来自crispr基因座的至少一个重复序列。在某些方面,引导rna的3’区还含有crrna和/或tracrrna的一个或多个结构序列。在一些实施方案中,引导rna包括crrna和tracrrna,这两条rna通过杂交形成复合物。
在一些实施方案中,基因组编辑组合物还包括含有供体序列的供体质粒。可通过包括转染、磷酸钙-dna共沉淀、deae-葡聚糖介导的转染、聚凝胺介导的转染、电穿孔、显微注射、转导、细胞融合、脂质体融合、脂质转染、原生质体融合、逆转录病毒感染、使用基因枪、使用dna载体转运子和生物弹(例如,粒子轰击)在内的各种本领域已知的手段将供体质粒引入细胞中。
在一些实施方案中,供体序列侧翼连接有5’同源臂和3’同源臂,其中5’同源臂与位于基因组上插入位点上游的5’目标序列同源,3’同源臂与位于基因组上插入位点下游的3’目标序列同源,其中可通过同源定向修复在插入位点将供体序列插入基因组中。
在一些实施方案中,采用本领已知的任意方法评估供体序列的插入。例如,可以设计与位于5’同源臂上游的序列对应的5’引物和与供体序列中的一区域对应的3’引物来评估插入物的5’连接。类似地,可以设计与位于3’同源臂下游的序列对应的3’引物和与供体序列中的一区域对应的5’引物来评估插入物的3’连接。
在一些实施方案中,插入位点可以位于任意所需的位点,只要设计rna引导的核酸内切酶以影响在该位点处的切割即可。在一些实施方案中,插入位点位于目标基因座。在一些实施方案中,插入位点不是基因座。
在一些实施方案中,供体核酸为不存在于宿主细胞中的序列。在一些实施方案中,供体序列为存在于除了预定目标位点以外的位点处的核酸内切酶序列。在一些实施方案中,供体序列为编码序列。在一些实施方案中,供体序列为非编码序列。在一些实施方案中,供体序列为突变的基因座。
在一些实施方案中,供体序列的大小为大约1bp至大约100kp。在某些实施方案中,供体序列的大小在大约1bp与大约10bp之间、在大约10bp与大约50bp之间、在大约50bp与大约100bp之间、在大约100bp与大约500bp之间、在大约500bp与大约1kb之间、在大约1kb与大约10kb之间、在大约10kb与大约50kb之间、在大约50kb与大约100kb之间,或在大约100kb以上。
在一些实施方案中,供体序列为待插入染色体中的外源基因。在一些实施方案中,供体序列为经修饰的序列,所述经修饰的序列可替换目标位点处的外源序列。例如,供体序列可以是具有所需突变的基因,并且可以用于存在于替换染色体上的外源基因。在一些实施方案中,供体序列为调控元件。在一些实施方案中,供体序列为编码报道蛋白质和/或rna的标签或编码序列。在一些实施方案中,在框架中将供体序列插入目标基因的编码序列中,所述目标基因将允许融合蛋白的表达,所述融合蛋白包含融合至目标蛋白的n或c末端的外源序列。
在一些实施方案中,在细胞内切割本文所述的供体质粒,产生线性核酸。本文所述的线性核酸包含5’同源臂、供体序列和3’同源臂。换言之,供体序列与5’同源臂和3’同源臂侧翼连接。
在一些实施方案中,同源臂的长度为至少大约50bp,例如,长度为至少大约50bp、100bp、200bp、300bp、600bp、900bp、1kb、1.5kb、2kb、4kb、6kb、10kb、15kb和20kp中的任意一者。在一些实施方案中,同源臂的长度为至少300bp。在某些实施方案中,同源臂长度可以为大约50bp至大约2000bp、大约100bp至大约2000bp、大约150bp至大约2000bp、大约300bp至大约2000bp、大约300bp至大约1500bp、大约300bp至大约1000bp。在一些实施方案中,5’同源臂的长度与3’同源臂的长度相同。在一些实施方案中,5’同源臂的长度与3’同源臂的长度不同。
在一些实施方案中,5’同源臂与位于基因组上的插入位点上游的5’目标序列同源,3’同源臂与位于基因组上的插入位点(例如,dsb)下游的3’目标序列同源,从而允许同源定向修复。在一些实施方案中,5’和/或3’同源臂可与距插入位点(例如,dna切割位点)小于200bp的对应目标序列同源。在一些实施方案中,5’和/或3’同源臂可与距dna切割位点小于0bp的目标序列同源。在一些实施方案中,5’目标序列与3’目标序列之间的间隔小于200bp。
在一些实施方案中,在细胞内切割供体质粒(例如,通过识别质粒上的切割位点的rna引导的核酸内切酶),产生本文所述的线性核酸。例如,供体质粒可包含位于5’同源臂上游和3’同源臂下游的侧翼序列。宿主细胞的基因组序列中不存在一些实施方案中的这些侧翼序列,从而允许仅在供体质粒上进行切割。引导rna可识别5’侧翼序列和3’侧翼序列。然后,可相应地设计rna引导的核酸内切酶以在引导rna的指导下影响在5’侧翼序列和3’侧翼序列处进行切割,所述引导rna可释放线性核酸,但不影响宿主序列。侧翼序列可以,例如,为大约20至大约23bp。
在一些实施方案中,所述方法还包括将bcl抑制因子进入细胞中。在一些实施方案中,将bcl抑制因子以蛋白质的形式或以编码bcl抑制因子的信使rna(mrna)或cdna等核酸的形式引入真核细胞中。核酸可作为质粒或病毒载体等较大构建体的一部分输送,或者例如直接通过电穿孔、脂质囊泡、病毒转运蛋白和显微注射输送。在一些实施方案中,可通过包括转染、磷酸钙-dna共沉淀、deae-葡聚糖介导的转染、聚凝胺介导的转染、电穿孔、显微注射、转导、细胞融合、脂质体融合、脂质转染、原生质体融合、逆转录病毒感染、使用基因枪、使用dna载体转运子和生物弹(例如,粒子轰击)在内的各种本领域已知的手段将bcl抑制因子引入细胞中。在一些实施方案中,将bcl抑制因子直接加入至培养基中,然后再通过与具有bcl抑制因子的细胞接触引入细胞中。在一些实施方案中,将至少两个bcl抑制因子引入细胞中。
在一些实施方案中,将细胞凋亡调节因子(例如,bcl-xl)、基因组编辑组合物和bcl抑制因子同时引入细胞中。在一些实施方案中,在与其他部件不同的时间将三个组分中的至少一个引入细胞中。例如,先将所述bcl-xl引入细胞中,然后再引入基因组编辑组合物和bcl抑制因子。在一些实施方案中,先将所述bcl-xl引入细胞中,然后再引入bcl抑制因子和基因组编辑组合物。在一些实施方案中,在彼此不同的时间点引入所有三个组分。例如,可以按特定的顺序依次施用这三个组分。在一些实施方案中,先将bcl-xl和基因组编辑组合物引入细胞中,然后再引入bcl抑制因子。
在一些实施方案中,将bcl抑制因子引入细胞中,以使得细胞中bcl抑制因子含量比对照细胞中bcl抑制因子的背景含量至少高20%、至少50%、至少75%、至少1倍、至少2倍或超过2倍。例如,在某些情况下,将bcl抑制因子引入细胞中,以使得细胞中bcl抑制因子的含量比对照细胞中bcl抑制因子的背景含量高25%至50%、50%至75%、75%至1倍,或者1倍至2倍。
在一些实施方案中,先将bcl抑制因子直接加入培养基中,然后再通过与含bcl抑制因子的细胞接触引入细胞中。培养基中bcl抑制因子的浓度的范围为0.5μm至1μm。在一些实施方案中,bcl抑制因子包括abt-263。
下面将参考实施例更详细地描述上述实施方案。然而,这些实施例不应视为在任何意义上限制本发明的范围。
实施例1:稳定的bcl-xl过表达
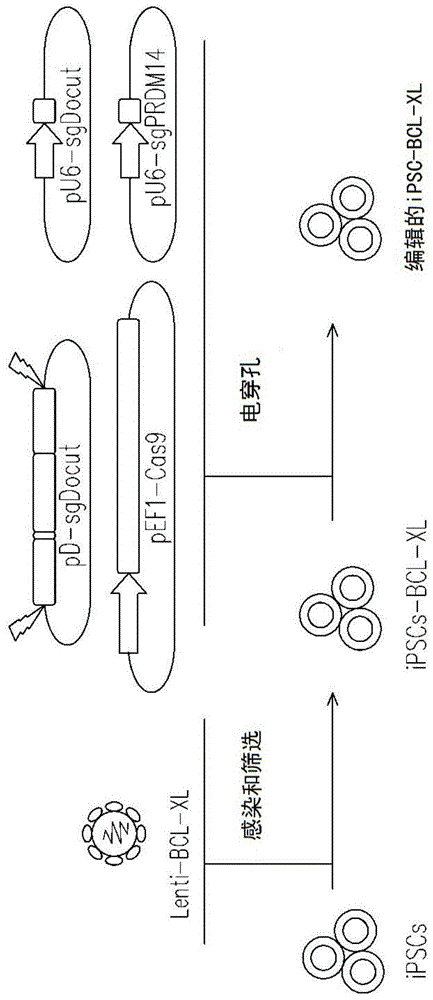
图1示出了人ipsc中稳定bcl-cl过表达的方案设计。在本实验中,用ipsc来检验稳定的bcl-xl过表达对编辑效率的影响。为实现该目的,建立ipsc-lenti-bcl-xl细胞(图1)。
按下面步骤构建在puro与wpre元件之间含有bcl-xl序列的慢性毒载体(lenti-ef1-bcl-xl-e2a-puro-wpre)。通过pcr扩增抗嘌呤霉素基因(puro)与bcl-xl的互补dna(cdna)并分别用kapahifi聚合酶(kapa生物系统)和genejet凝胶提取试剂盒(thermofisherscientific)进行纯化。用nebuilder-hifi-dna组装试剂盒(新英格兰生物实验室)将bcl-xl、e2a连接物和puro的片段插入带有ef1启动子的慢病毒载体中。所有构建体均经sanger测序(mclab)来验证。在circlegrow培养基(mp生物医学)中培养正确的克隆,并用无内切核酸酶质粒maxi试剂盒(qiagen)纯化dna质粒。标准的磷酸钙沉淀协议用于慢病毒生产。在4℃下以6000g离心24小时,慢病毒载体被浓缩100倍,达到2-10x107/ml的生物效价。
用慢病毒载体(lenti-ef1-bcl-xl-e2a-puro-wpre)在0.1-0.2的低moi条件下转导ipsc,稳定转导的细胞通过将细胞在补充有1μg/ml嘌呤霉素的mtesr1培养基中培养1周来选择。抗生素筛选一周后,建立表达bcl-xl的ipsc-bcl-xl细胞系。作为对照,还可仅通过用抗puro基因进行转导来建立ipsc-bcl-xl对照细胞系。
通过创建pdrm14的绿色荧光蛋白报告子系来检测稳定bcl-xl过表达的效果。图2是在pdrm14处进行hdr编辑的示意图。构建以下质粒:pef1-cas9、pu6-sgprdm14、pd-prdm14-e2a-mneongreen-sg和pu6-sgdocut。
用nebuilderhifi组装试剂盒(新英格兰生物实验室)构建所有的cas9plasmid(pef1-cas9)和sgrnaplasmid(pu6-sgprdm14和pu6-sgdocut)。首先,用kapahifi聚合酶(kapa生物系)生产pcr产物并用genejet凝胶提取试剂盒(thermofisherscientific)纯化。然后,根据制造商的说明,在冰上,在dna组装反应(20μl)将线性pcr产物组装为质粒。该反应中含有nebuilderhifidna组装主混合物(10μl),等比例的pcr产物和水。在50℃孵育5~30min之前,简短地涡旋连接反应并离心。然后用组装的dna产物转化neb5-alpha-感受态大肠杆菌细胞,并用氨苄西林在lb琼脂平板上进行铺板。选择多个菌落进行sanger测序(mclab),确定正确的克隆。sgdocut序列为gggtgcgagatgaactca(seqidno:2)。sgprm14序列为gagactactagctcctgcc(seqidno:3)。设计sgprmd14以切割prdm14,设计sgdocut以切割双切供体质粒(pd-sg,即pd-prd14-e2amneongreen-sg)。
使用nebuilderhifidna组装试剂盒(新英格兰生物实验室)生成双切供体质粒(pd-sg,即pd-prd14-e2amneongreen-sg),如上详述。简言之,采用kapahifi聚合酶(kapa生物系)由pcr来扩增pdonor-sg(左同源臂,基因敲入所需的片段,右同源臂)中包含的所有片段并用genejet凝胶提取试剂盒(thermofisherscientific)纯化。从人gdna中扩增长度为~600bp的ha序列,将sgdocut识别序列添加于左ha的上游和右ha的下游。所有载体通过sanger测序来验证。
将包括pef1-cas9、pu6-sgprdm14、pd-prdm14-e2a-mneongreen-sg和pu6-sgdocut在内的crispr质粒共电穿孔为ipsc-lenti-bcl-xl系和ipsc-lenti-对照系,从而通过hdr途径将e2a-mneongreen精确整合在prdm14终止密码子处。对于人ipsc的电穿孔,根据制造商的说明,采用
实施例2:瞬时bcl-xl过表达
图4示出了人ipsc中瞬时bcl-cl过表达的方案设计。在该实验中,构建了在ef1启动子的控制下编码bcl-xl的质粒(pef1-bcl-xl)。采用上文详述的nebuilderhifidna组装试剂盒(新英格兰生物实验室)生成pef1-bcl-xl质粒。
通过电穿孔将crispr质粒(pef1-cas9、pu6-sgprdm14、pd-prdm14-e2a-mneongreen-sg和pu6-sgdocut)与pef1-bcl-xl质粒一起共转染为ipsc(图4)。因为在哺乳动物细胞中细胞分裂和逐步沉默后载体缺失,所以载体仅提供瞬时bcl-xl过表达。作为对照,也可通过仅用crispr质粒(无需bcl-xl)进行转导来建立对照组。
图5示出了瞬时bcl-xl过表达组和对照组的流式细胞术分析。如上详细所述,检测第一天的细胞存活率和第3天的hdr基因敲入(ki)效率。与无bcl-xl的对照组中3%的细胞存活率相比,瞬时bcl-xl过表达组中有27%的细胞存活,增加了9倍(图5)。存活率增加导致prdm14基因座处的ki效率(0.17%vs.25%)提高大约150倍(图5)。应注意的是,与稳定的bcl-xl过表达(25%vs.15%)相比,瞬时bcl-xl体现了更高的编辑效率(图5、图3)。这可能是由于瞬时转染bcl-xl后选择压力增大所致,因为转染有更多bcl-xl拷贝数和编辑质粒的细胞更容易在压力下存活并具有更高的编辑效率。
实施例3:bcl-xl对ipsc中ctnnb1或oct4基因座处的ipsc存活率和hdr效率的影响
为了概括bcl-xl对ipsc存活率和hdr介导的ki的影响,我们在另外两个基因座ctnnb1和oct4处进行了类似的实验。图6是在ctnnb1处进行hdr编辑的示意图。图7是在oct4处进行hdr编辑的示意图。构建了以下质粒:pu6-sgctnnb1、pd-ctnnb1-e2a-mneongreen-sg、pu6-sgoct4和pd-oct4-e2a-puro-e2a-crimson-sg。
ctnnb1是典型wnt途径中的关键基因,其组成性地表达在ipsc和其他细胞中。用pu6-sgctnnb1和pd-ctnnb1-e2a-mneongreen-sg在终止密码子前的39bp处进行靶向,并将e2a-mneongreen盒插入ctnnb1基因座a。sgctnnb1序列为gctgattgctgtcactggg(seqidno:4)。
在双切hdr供体pd-oct-e2apuro-e2acrimson-sg的oct4终止密码子前框内插入e2a-puro-e2a-crimson盒,左ha(600bp)从内含子2延伸到外显子4和右ha(1000bp)从终止密码子开始,从而导致了可用pdonor上的外显子5-e2a-puro-e2a-crimson替换基因组上的内含子4和外显子5。标靶oct4内含子4的sgoct4序列为gtgagtgccatgtctctctg(seqidno:5)。
在本实验中,使用上文详述的nebuilderhifidna组装试剂盒(新英格兰生物实验室)生成本实验中使用的所有sgrna质粒和供体质粒。为了进一步增加重复性,本实验使用了6种不同的ipsc系。
crispr质粒(pef1-cas9、pu6-sgctnnb1、pd-ctnnb1-e2a-mneongreen-sg和pu6-sgdocut)和crispr质粒(pef1-cas9、pu6-sgoct4、pd-oct4-e2a-puro-e2a-crimson-sg和pu6-sgdocut)以及pef1bcl-xl质粒通过电穿孔分别共转染为ipsc。在无启动子pdonor载体的hdr基因敲入后,荧光报告因子mneongreen或crimson的表达将受到内源性ctnnb1或oct4转录机制的驱动。
图8示出了指示ipsc中ctnnb1和oct4处的ki效率的流式细胞术分析。如上详述,在第3天检测hdr基因敲入(ki)效率。同样,bcl-xl共转染导致ctnnb1(1.6%vs.32%)和oct4(0.4%vs.7.6%)的ki效率均显著提高20倍(图8)。在上述基础上,bcl-xl可提高电穿孔后应激ipsc的存活率。
实施例4:bcl-xl对ipsc中ctnnb1和oct4处的基因敲除效率的影响
图9是c326处的nhej介导的基因敲除的示意图。图10是cd9处的nhej介导的基因敲除的示意图。构建以下质粒:pu6-sgcd326和pu6-sgcd9。设计sgcd9以靶向cd326的外显子1。sgcd326序列为ggttctcactcgctcagac(seqidno:6)。设计sgcd9以靶向cd9的外显子2。sgcd9序列为gcttctacaggtagggga(seqidno:7)。
通过电穿孔法分别将crispr质粒(pef1-cas9和pu6-sgcd326)和crispr质粒(pef1-cas9和pu6-sgcd9)与pef1-bcl-xl质粒共转染为ipsc。
这两个基因的敲除是因为它们可在人ipsc中高水平表达,并且可以用表面标记的抗体(抗人cd9futc(ebioscience)和抗人cd326pe(ebioscience))染色和流式细胞术来检测。用抗cd326pe染色和门控pe阴性细胞在电穿孔后1周确定cd326基因敲除效率。用抗cd9fitc染色法和门控fitc阴性细胞在电穿孔后1周确定6个ipsc细胞系中的cd9基因敲除效率。
图11示出了指示ipsc中cd326和cd9处的ko效率的流式细胞术分析。通过bcl-xl可显著提高ko效率,cd326处提高5.5倍(10%vs.55%),cd9处提高2.5倍(9%vs.22%)。瞬时bcl-xl过表达可显著提高hdr基因敲入效率和nhej介导的基因敲除效率。
实施例5:在存在或不存在bcl-xl的条件下电穿孔后细胞死亡的动态分析
在本实验中,对转染后2-48小时的活细胞计数以研究在不存在或存在bcl-xl的情况下对使用质粒进行ipsc电穿孔后的细胞死亡的动力学。
图12示出了用基因组编辑质粒结合或不结合bcl-xl电穿孔后相对细胞数的动态变化。电穿孔后约0.5h,两组中均有约50%的ipsc幸免于电穿孔的冲击。0.5小时到2小时,来自两组的细胞附着更加稳定,没有观察到有额外的细胞死亡(图12)。然而,从2小时到9小时,对照组中大部分的细胞开始死亡。相反地,bcl-xl的不断表达开始保护细胞免于死亡,这导致了在第4小时时的存活率增加60%。6小时后,随着bcl-xl的积聚,此后细胞死亡没有显著增加。在用bcl-xl进行电穿孔后24小时,细胞开始分裂,与无bcl-xl对照组相比,导致相对存活率从大约10倍增加到大约20倍(图12)。这些结果证实了电穿孔后2至8小时细胞快速死亡,而过表达的bcl-xl可防止细胞在电穿孔后4小时开始死亡。
实施例6:bcl2家族对细胞存活率和编辑的影响
在本实验中,构建了pef1-bcl2和pef1-mcl1质粒以与pef1-bcl-xl质粒比较。如上详述,用nebuilderhifidna组装试剂盒(新英格兰生物实验室)产生pef1-bcl2和pef1-mcl1质粒。
用与上述实施例2和实施例4相似的方法在hdr基因敲入系统和nhej基因敲除系统中测试pef1-bcl-xl、pef1-bcl2、pef1-mcl1质粒。作为对照,对照组也可通过仅用crispr质粒进行转导来建立。电穿孔后1天,通过细胞计数来确定细胞存活率。通过facs在3或7天确定ki或ko效率。
图13示出了结合bcl-xl、bcl2或mcl1的ipsc的流式细胞术分析。在这两个系统(hdr基因敲入和nhej基因敲除)中,各个载体的细胞存活率没有区别,因此可组合在一起进行分析。bcl2也提高了存活率(约5倍),但与bcl-xl(约8倍)相比处于较低水平,而mcl1在质粒转染后并没有显著提高ipsc存活率。相应地,bcl2适度增加了hdr介导的ki效率(0.2%vs.7.5%,提高了约35倍)和nhej介导的ko效率(9.2%vs.37%,提高了约4倍),而mcl1则均未显著提高ki和ko效率(图13)。这些数据表明,bcl-xl的显著作用不能被bcl2或mcl1所替代。换言之,bcl2或mcl1在ipsc存活率和编辑方面低于bcl-xl。
此外,通过rt-pcr测定转染后8小时cas9mrna和sgrna在ipsc中的表达。
图14示出了cas9和sgrna的表达水平。人们观察到,cas9表达水平增加了5-10倍,sgrna表达水平增加了3-6倍。这些数据表明,bcl-xl或bcl2优先保护转染有更多质粒拷贝数的细胞免于死亡,从而导致存活的ipsc中crispr组分的含量更高,进而提高编辑效率。
实施例7:bcl-xl对bax基因敲除细胞系的影响
bcl-xl与促凋亡对应物bax和bak1结合,阻止了线粒体外膜中致死孔的形成,从而中断细胞凋亡。人ipsc主要表达bax而非bak1(约300vs.约50tpm或百万分之转录本)(未示出)。在本实验中,通过用cas9-sgrna靶向bax外显子1或外显子2,在两个不同的ipsc细胞系中建立了4个baxko细胞系。以野生型ipsc作为对照。
设计靶向bax的sgrna(sgbax-e1、sgbax-e2)。并设计和构建pd-ef1-puro-polya-sg。用cas9、sgrna(靶向bax)、pd-ef1-puro-polya-sg和bcl-xl的质粒电穿孔两个人ipsc细胞系。将1μg/ml嘌呤霉素加入培养基中1周,筛选出稳定的bax基因敲除细胞系。sgbax-e1序列为gcggcggtgatgacgggtc(seqidno:8)。sgbax-e2序列为gacagggcccttttgcttc(seqidno:9)。
图15示出了bax基因敲除对ipsc细胞存活率和编辑效率的影响。这四个系中的baxko大大提高了pdrm14和ctnnb1基因座处的细胞存活率和hdr介导的ki(图15)。然而,bcl-xl的添加显著增加了hdr效率,表明了baxko不能单独替代bcl-xl。
实施例8:bcl-xl对bbc3基因敲除细胞系的影响
bbc3,又称puma,与bcl2家族成员相互作用,从而释放出bax或bak1并诱导细胞凋亡。在本实验中,通过用cas9-sgrna靶向bbc3外显子1或2,在两个不同的ipsc细胞系中建立4个bbc3ko细胞系。
设计靶向bbc的sgrna(sgbbc3-e1、sgbbc3-e2)。并设计和构建pd-ef1-puro-polya-sg。用cas9、sgrna(靶向bbc3)、pd-ef1-puro-polya-sg和bcl-xl质粒电穿孔两个人ipsc细胞系。将1μg/ml嘌呤霉素加入培养基中1周,筛选出稳定的bax基因敲除细胞系。sgbbc3-e1序列为gactcacaaatctggca(seqidno:10)。sgbbc3-e2序列为gtaggggcctggcccga(seqidno:11)。
图16示出了bbc3基因敲除对ipsc细胞存活率和编辑效率的影响。与baxko相似,bbc3ko显著提高了细胞存活率和编辑,但bcl-xl的添加进一步增加了两基因座处的hdr效率(图16),表明了bbc3缺失也不能取代bcl-xl。
实施例9:bcl抑制因子abt-263对ipsc编辑的影响
含有高拷贝数crispr质粒的ipsc可以提高编辑效率,从而富集这些细胞可以增加较大群体的编辑效率。为了优先去除转染有低拷贝质粒的细胞,用abt-263(navitoclax)、bcl-xl、bcl-2和bcl-w的有效抑制因子来处理细胞。
为了测试abt-263的作用,在用crispr质粒(pef1-cas9、pu6-sgprdm14、pd-prdm14-e2a-mneongreen-sg和pu6-sgdocut)和pef1-bcl-xl进行电穿孔后将ipsc均分为若干孔。首先,在50μl培养基中稀释bcl抑制因子abt-263(navitoclax;selleckchemicals),制备主混合物,然后以所需的工作浓度和处理时间将50μl稀释的小分子均匀地加入到每个孔中。此后,用新鲜培养基更换该培养基。作为对照,平行孔中仅添加dmso(0.1%)。电穿孔后3天,收集细胞进行facs分析,确定在每种条件下的细胞存活率和编辑效率。
图17示出了abt-263的不同剂量和治疗周期对电穿孔后的ipsc细胞存活率的影响。图18示出了电穿孔后不同剂量和治疗周期的abt-263对ipsc的hdr效率的影响。
由图17和图18可知,当没有外源性bcl-xl表达时,电穿孔后即刻加入abt-263(0.2μm,0.5μm)会导致细胞存活率急剧下降,编辑效率无显著提高。相反地,当从8小时开始施用abt-263时,当稳健的bcl-xl被表达直到24小时时,才观察到细胞存活率剂量依赖性逐渐下降和编辑效率提高。
此外,还测试了abt-263对ko效率的影响。采用与实施例4类似的方法确定ko效率。图19示出了abt-263对ipsc细胞存活率和编辑效率的影响。从图17、18、19可知,在1μm的abt-263条件下,细胞存活率降低50-70%,ki和ko编辑效率分别提高70%和40%。这些结果表明,在ipsc基因组编辑中使用bcl-xl可允许通过施用bcl抑制因子进行细胞筛选和富集成功编辑的细胞。
实施例10:经双或等位编辑的快速高水平基因敲入或基因敲除
图20是ipsc中prdm14和ctnnb1处的hdr介导的双重ki的示意图。在本实验中,设计具有抗嘌呤霉素基因的双切供体(pd-e2a-puro-e2a-crimson-sg)来靶向pdrm14基因座。将crispr质粒(pd-e2a-puro-e2a-crimson-sg、pd-ctnnb1-e2a-mneongreen-sg、pu6-sgctnnb1、pu6-sgprdm14、pef1-cas9、pu6-sgdocut)与bcl-xl质粒共电穿孔为ipsc。通过prdm14处的hdr基因敲入进行的e2a-puro-e2a-crimson盒的基因敲入允许选择嘌呤霉素以在ctnnb1处用hdr编辑来富集ipsc。具体来说,在电穿孔后2天添加1μg/ml的嘌呤霉素进行筛选。用facs确定ctnnb1处的ki效率(mneongreen阳性)。
图21示出了指示ipsc中ctnnb1处的ki效率的流式细胞术分析。由图21可知,经嘌呤霉素筛选后,ctnnb1处的hdrki效率从17%增加到95%。
图22是由hdr在prdm14处和由nhej在cd326处进行双重编辑的示意图。将crispr质粒(pd-e2a-puro-e2a-crimson-sg、pu6-sgcd326、pu6-sgprdm14、pef1-cas9、pu6-sgdocut)与bcl-xl质粒共电穿孔为ipsc。在prdm14处通过hdr基因敲入e2a-puro-e2a-crimson盒来选择嘌呤霉素以在cd326处用nhej编辑富集ipsc。用facs确定cd326(cd326pe阴性)处的ko效率。
图23示出了指示ipsc中cd326处ko效率的流式细胞术分析。从图23可以知道,含有cd326ko的细胞从21%富集到98%。这些数据表明,在批量ipsc中,采用双重编辑策略以易于将hdr或ko编辑效率提高到95%以上。
图24是通过片段盒的双等位hdr插入来进行基因敲除的示意图。在本实验中,以600bp同源臂设计两个双切hdr供体(pd-ef1-puro-sg和pd-ef1-zeocin-sg)以在cd326处插入抗嘌呤霉素或博来霉素基因,从而导致开放阅读框的双等位基因断裂。
将所有crispr质粒(pd-ef1-puro-sg、pd-ef1-zeocin-sg、pu6-sgcd326、pef1-cas9、pu6-sgdocut)与bcl-xl质粒共电穿孔为ipsc。电穿孔后1周,进行抗体染色和facs分析。图25示出了指示ipsc中cd326处的ko效率的流式细胞术分析。从图25可知,观察到ko效率为38%。用嘌呤霉素或佐西林进行选择可将ko效率提高到95-96%,而用嘌呤霉素和佐西林进行双重选择可使ko效率达到100%(图25)。这些数据表明,通过选择性盒的双等位基因敲除来实现几乎100%的细胞中的基因敲除。
实施例11:bcl2家族对多个细胞系的编辑的不同影响
在本实验检,测试了小鼠escs、293t(人胚肾细胞)、k562(人红白血病细胞)和jurkat(t细胞白血病)细胞中bcl-xl、bcl2和mcl1的作用。在所有这些细胞系中,用bcl进行电穿孔并没有增加细胞存活率(数据未显示)。
为了便于检测hdr编辑,设计无启动子的pdonor-mneongreen以在通用表达基因eef1a1和gapdh的终止密码子定位的外显子之前靶向内含子。在小鼠esc中,设计两个sgrna(sgeef1a1-1、sgeef1a1-2)以靶向eef1a1内含子。sgeef1a1-1序列为gagttagcagactcagatc(seqidno:12)。sgeef1a1-2序列为gtagcaaagatactgataaa(seqidno:13)。此外,在293t、k562和jurkat细胞中,设计sgeef1a1-1和sggapdh以靶向eef1a1和gapdh内含子。sgeef1a1-1序列为gtagtcctctatcccaa(seqidno:14)。sggapdh序列为gacaacttttcattct(seqidno:15)。
从atcc购买小鼠es细胞并将其维持在杜尔贝科氏改良eagle培养基(dmem)中,该培养基补充有5%胎牛血清(gibco)、5%血清替代物(gibco)、2mm谷氨酰胺(gibco)、10ng/ml小鼠白血病抑制因子(lif)、0.1mm2-巯基乙醇(gibco)、3μmgsk抑制因子(chir99021,selleck)、1μmmek抑制因子
在含10%fbs(gibco)和1%青霉素/链霉素(invitrogen)的rpmi-1640(vwr生命科学)中生长k562(atcc,ccl-243)细胞。为了电穿孔k562细胞,按照制造商的说明,使用氨甲喋呤细胞系nucleofectortm试剂盒v(lonza)和程序t-016。
在含10%fbs(gibco)和1%青霉素/链霉素(invitrogen)的rpmi-1640(vwr生命科学)中生长jurkat(atcc,克隆e6-1)细胞。为了电穿孔jurkat细胞,根据制造商的说明,使用amaxatm细胞系nucleofectortm试剂盒v(lonza)和程序x-001。
在补充有10%胎牛血清(fbs;gibco)和1%青霉素/链霉素(invitrogen)的dmem(杜尔贝科氏改良eagle培养基,sigma)中培养hek293t细胞。为了转染293t细胞,根据制造商的说明使用lipofectamine3000(生命技术)。
在存在或不存在bcl质粒的条件下,用编辑质粒电穿孔小鼠esc、k562、293t细胞。通过脂质体转染法转染293t细胞。在通过facs进行转染后3天确定经部分mneongreen阳性细胞反映的hdr基因敲入效率。
图26示出了瞬时bcl-xl、bcl2或mcl1过表达对小鼠es细胞中hdr效率的影响。在2个小鼠esc实验中,bcl-xl显著增加了hdr编辑,而bcl2和mcl1的效果较差。
图27示出了瞬时bcl-xl、bcl2或mcl1过表达对k562细胞中hdr效率的影响。在k562细胞中,bcl-xl和mcl1没有明显益处,而bcl2则均显著降低了eef1a1和gapdh处的hdr效率。
图28示出了瞬时bcl-xl、bcl2或mcl1过表达对293t细胞中hdr效率的影响。在293t细胞中,bcl-xl和bcl2均显著降低了eef1a1和gapdh处的hdr效率,而mcl1则无明显效果。
图29示出了瞬时bcl-xl、bcl2或mcl1过表达对jurkat细胞中hdr效率的影响。在jurkat细胞中,bcl-xl和mcl1有提高编辑效率的趋势,但差异不显著。
综上所述,这些数据表明,bcl-xl增加了esc中的hdr效率,通过影响细胞存活率以外的机制,bcl2成员对不同细胞系中的基因编辑有不同的影响。
对本领域技术人员来说,显而易见的是,在不脱离本发明的范围或精神的情况下,可以对所公开的实施方案进行各种修改和变化。鉴于上述情况,本发明旨在涵盖修改和变化,只要它们都包含在下述权利要求书及其等同替换的范围内。
序列表
<110>中国医学科学院血液病医院(中国医学科学院血液学研究所)
<120>在哺乳动物干细胞中进行基因组编辑的方法
<130>xys19005-1
<160>15
<170>siposequencelisting1.0
<210>1
<211>233
<212>prt
<213>人类(human)
<400>1
metserglnserasnarggluleuvalvalasppheleusertyrlys
151015
leuserglnlysglytyrsertrpserglnpheseraspvalgluglu
202530
asnargthrglualaprogluglythrgluserglumetgluthrpro
354045
seralaileasnglyasnprosertrphisleualaaspserproala
505560
valasnglyalathrglyhisserserserleuaspalaarggluval
65707580
ileprometalaalavallysglnalaleuargglualaglyaspglu
859095
phegluleuargtyrargargalapheseraspleuthrserglnleu
100105110
hisilethrproglythralatyrglnserphegluglnvalvalasn
115120125
gluleupheargaspglyvalasntrpglyargilevalalaphephe
130135140
serpheglyglyalaleucysvalgluservalasplysglumetgln
145150155160
valleuvalserargilealaalatrpmetalathrtyrleuasnasp
165170175
hisleugluprotrpileglngluasnglyglytrpaspthrpheval
180185190
gluleutyrglyasnasnalaalaalagluserarglysglyglnglu
195200205
argpheasnargtrppheleuthrglymetthrvalalaglyvalval
210215220
leuleuglyserleupheserarglys
225230
<210>2
<211>18
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(18)
<223>合成寡核苷酸
<400>2
gggtgcagatgaacttca18
<210>3
<211>19
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(19)
<223>合成寡核苷酸
<400>3
gaagactactagccctgcc19
<210>4
<211>19
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(19)
<223>合成寡核苷酸
<400>4
gctgattgctgtcacctgg19
<210>5
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>5
gtgagtgccatgtctctctg20
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>6
ggttctcactcgctcagagc20
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>7
gcttctacacaggtgaggga20
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>8
gcggcggtgatggacgggtc20
<210>9
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>9
gacaggggcccttttgcttc20
<210>10
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>10
gactcaccacaaatctggca20
<210>11
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>11
gtagagggcctggcccgcga20
<210>12
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>12
gagtgtagcagactcagatc20
<210>13
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>13
gtagcaaagatactgataaa20
<210>14
<211>18
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(18)
<223>合成寡核苷酸
<400>14
gtagtcatccttacccaa18
<210>15
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<221>misc_feature
<222>(1)..(20)
<223>合成寡核苷酸
<400>15
gacaactcttttcatcttct20
- 还没有人留言评论。精彩留言会获得点赞!