一种合成四氢吲唑化合物的方法与流程
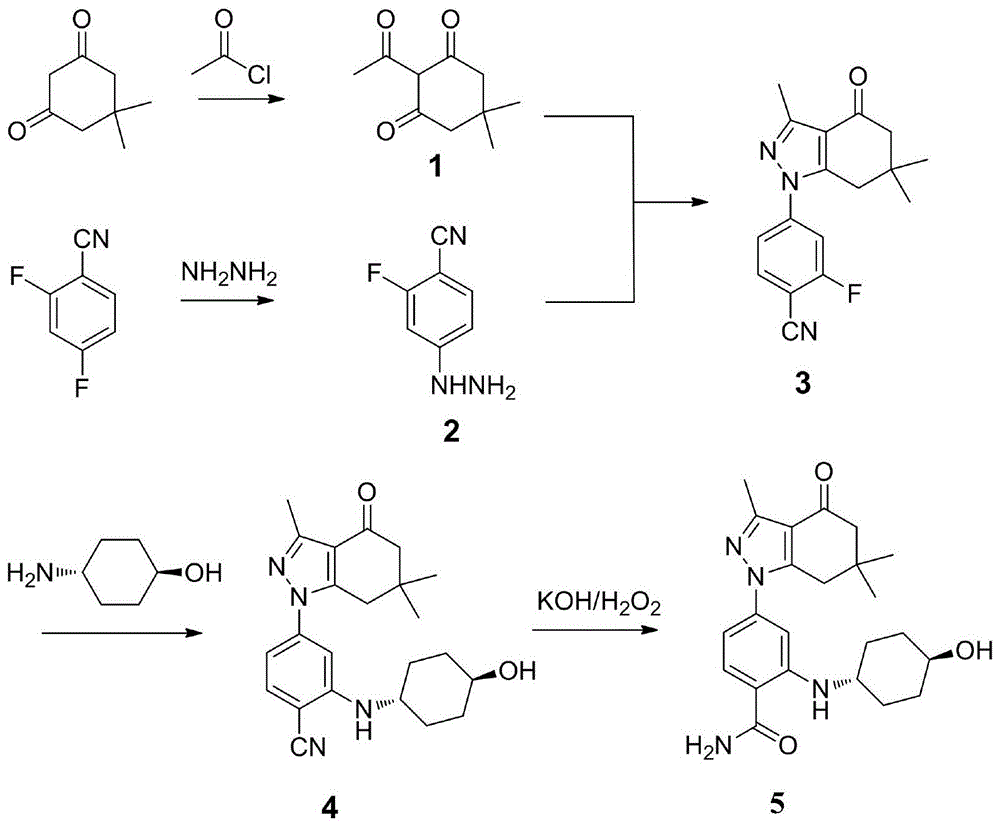
本发明属于药物合成
技术领域:
,具体涉及一种合成四氢吲唑化合物的方法。
背景技术:
:四氢吲唑化合物是一种新型、具有很好前景的抗病毒药物。它对多种病毒,尤其对疱疹病毒i和ii型,柯萨奇病毒iii型(cvb3)有明显的抑制作用,专利文献cn101273991a公开了四氢吲哚酮/四氢吲唑酮/四氢咔唑衍生物及其盐在制备抗病毒药物中的应用,说明四氢吲唑酮化合物对乙肝病毒等具有很好的活性。此外,专利文献cn1896060a公开了四氢吲哚酮的衍生物和四氢吲唑酮的衍生物及它们的应用,同时还提到了四氢吲唑化合物在抗肿瘤方面也具有潜在的应用价值,鉴于四氢吲唑化合物的多种活性作用,市场对四氢吲唑化合物的需求量也日益增加。现有技术中,四氢吲唑化合物的合成采取了线性方法,即通过5,5-二甲基-1,3-环己二酮和乙酰氯反应得到5,5-二甲基-2-乙酰基-1,3-环己二酮,再和水合肼反应得到3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑,接着和2,4-二氟苯甲腈反应得到2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈,然后和反式4-羟基环己胺反应得到2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈,最后和koh/过氧化氢反应得到四氢吲唑化合物。此工艺的最大缺点是在制备2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的步骤中同时生产约40%的4-氟-2-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的副产物,而且此副产物和主产物难以分开,需要采用硅胶柱层析的方法进行纯化。因此,该工艺路线存在成本高,至少浪费了一半的3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑和2,4-二氟苯甲腈。因此,为满足市场需求,研究开发一条简单的、低成本的、能够放大生产四氢吲唑化合物的合成路线具有必要性。技术实现要素:本发明旨在提供一种合成四氢吲唑化合物的方法,该合成方法中的中间体3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑和2,4-二氟苯甲腈的合成率高,使最终合成的四氢吲唑化合物产率曾高,节约资源,适合大量生产。为了达到上述目的,本发明采用以下技术方案:本发明提供了一种合成四氢吲唑化合物的方法,合成方法包括以下步骤:s1:5,5-二甲基-2-乙酰基-1,3-环己二酮的合成:称取5,5-二甲基-1,3-环己二酮加入有机溶剂中溶解,再加入三乙胺和4-二甲氨基吡啶,0℃~4℃冰浴条件下,滴加乙酰氯,置于0℃~60℃条件下反应2.5-3.5h,过滤,取有机层,加入饱和食盐水清洗后,再加入无水na2so4干燥,减压除去溶剂,柱层析纯化,即得;s2:2-氟-4-肼基苯甲腈的合成:称取2,4-二氟苯甲腈加入有机溶剂中溶解,再加入水合肼,20℃~80℃条件下搅拌反应10-14h,减压旋干,加入饱和食盐水清洗后,再加入无水na2so4干燥,减压除去溶剂,加入乙酸乙酯重结晶,即得;s3:2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的合成:称取步骤s1得到的化合物5,5-二甲基-2-乙酰基-1,3-环己二酮和步骤s2得到的化合物2-氟-4-肼基苯甲腈加入有机溶剂中,再加入冰乙酸,25℃~80℃条件下反应6-12h,减压去除溶剂,加入乙酸乙酯萃取,先加入饱和nahco3清洗,再加入饱和食盐水洗涤,重复清洗2-3次,再加入无水na2so4干燥,浓缩溶剂,加入乙醇重结晶,即得;s4:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的合成:称取反式4-羟基环己胺,将其和步骤s3得到的化合物2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈加至二甲基亚砜中,加入k2co3,在25℃~80℃条件下反应10-12h后,冷却至室温,将反应液加入蒸馏水中,过滤,取滤渣,加入乙醇重结晶,即得;s5:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲酰胺的合成:称取步骤s4得到的化合物2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈溶于二甲基亚砜中,再加入koh和30%(v/v)的h2o2溶液,在室温条件下反应2h后,将反应液加入蒸馏水中,过滤,取滤渣,加乙醇重结晶,即得四氢吲唑化合物:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲酰胺。进一步地,所述的步骤s1中三乙胺、4-二甲氨基吡啶和乙酰氯与5,5-二甲基-1,3-环己二酮的摩尔比为(1.5-2):(0.25-0.5):(1.05-1.525):1。进一步地,所述步骤s1中的有机溶剂为乙腈、二氧六环或乙酸乙酯中的一种。进一步地,所述步骤s2中的水合肼与2,4-二氟苯甲腈的摩尔比为(1.0-2):1。进一步地,所述步骤s2中的有机溶剂为甲醇、乙醇、苯甲醇、乙二醇、二氯甲烷、二氧六环中的一种。进一步地,所述5,5-二甲基-2-乙酰基-1,3-环己二酮与2-氟-4-肼基苯甲腈的摩尔比为1:1;所述冰乙酸与5,5-二甲基-2-乙酰基-1,3-环己二酮的摩尔比为(1.0-1.5):1;所述步骤s3中的有机溶剂为甲醇、乙醇和二氧六环中的一种。进一步地,所述步骤s4中反式4-羟基环己胺和k2co3与2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的摩尔比为2:4.35:1。进一步地,所述步骤s5中的koh和30%(v/v)的h2o2溶液与2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的摩尔比为(0.012-0.048):1:1。更进一步地,所述步骤s1中的反应温度为20℃~35℃;所述步骤s2中的反应温度为50℃~80℃;所述步骤s3中反应温度为60℃~80℃;所述步骤s4中反应温度为60℃~80℃。本发明提供的合成四氢吲唑化合物的方法中2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈产率为74%-93%,该产率是现有技术的1.5-2.5倍,合成过程中的副产物生产量少,本发明提供的合成路线中还可将2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈与副产物4-氟-2-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈分离完全,提高纯度。另外中间物5,5-二甲基-2-乙酰基-1,3-环己二酮的产率为78%-95%;2-氟-4-肼基苯甲腈产率为52%-63%;2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈产率为65%-97%;2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲酰胺产率为83%-95%,各中间产物的产率高,优选出的合成路线成本低,可供大批量生产。与现有技术相比,本发明提供的合成四氢吲唑化合物的方法具有以下优点:(1)本发明提供的合成四氢吲唑化合物的方法的中间物2-氟-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈的产率高达93%,可与副产物4-氟-2-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈分离完全,产品纯度高。(2)本发明提供的四氢吲唑化合物的合成方法减少了副产物的生成,制备得到的四氢吲唑化合物纯度高,合成方法快速有效,合成成本低,适合大批量生产。附图说明图1为四氢吲唑化合物的合成路线,图中1为:5,5-二甲基-2-乙酰基1,3-环己二酮;2为:2-氟-4-肼基苯甲腈;3为:2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈;4为:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈;5为:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲酰胺。具体实施方式以下通过实施例形式的具体实施方式,对本发明的上述内容作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下实施例。实施例1、合成四氢吲唑化合物的方法s1:5,5-二甲基-2-乙酰基1,3-环己二酮的合成:称取20.0mmol的5,5-二甲基-1,3-环己二酮溶于10ml乙腈中,加入30.0mmol的三乙胺和5.0mmol的4-二甲氨基吡啶,0℃~4℃冰浴条件下滴加25.0mmol的乙酰氯,置于25℃条件下反应3h,过滤,取有机层,加入饱和食盐水清洗,再加入无水na2so4干燥,减压除去溶剂,柱层析纯化得到淡黄色油状液体1.4g;s2:2-氟-4-肼基苯甲腈的合成:称取7.19mmol的2,4-二氟苯甲腈加入10ml二氯甲烷中溶解,再加入7.19mmol的水合肼,25℃条件下搅拌反应10h,减压旋干,加入饱和食盐水清洗,再加入无水na2so4干燥,减压除去溶剂,加入甲醇重结晶得到白色固体产物0.56g,熔点137-138℃;s3:2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的合成:称取9.9mmol步骤s1得到的化合物5,5-二甲基-2-乙酰基-1,3-环己二酮和9.9mmol步骤s2得到的化合物2-氟-4-肼基苯甲腈加入30ml甲醇中,再加入10mmol冰乙酸,25℃条件下反应12h,减压去除溶剂,用乙酸乙酯萃取,先加入饱和nahco3清洗,再加入饱和食盐水洗涤,重复清洗和洗涤2-3次,再加入无水na2so4干燥,浓缩溶剂,加入乙醇重结晶,得到白色固体产物2.2g,熔点150-151℃;s4:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的合成:称取20mmol的4-羟基环己胺和10mmol步骤s3制得的2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈加至40ml二甲基亚砜中,加入43.5mmol的k2co3,在50℃条件下反应10h,冷却至室温,将反应液加入150ml蒸馏水中,过滤,取滤渣,加入乙醇重结晶,得到固体产物3.0g,熔点191-192℃;s5:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲酰胺的合成:称取2.5mmol步骤s4得到的化合物2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈溶于10ml二甲基亚砜中,再加入1.2mmol的koh和2.5mmol的30%(v/v)的h2o2溶液,在25℃条件下反应2h后,将反应液加入蒸馏水中,过滤,取滤渣,加乙醇重结晶得到产物0.87g,熔点220-221.0℃。合成路线图见图1。实施例2、合成四氢吲唑化合物的方法s1:5,5-二甲基-2-乙酰基1,3-环己二酮的合成:称取20.0mmol的5,5-二甲基-1,3-环己二酮溶于20ml二氧六环中,加入30.0mmol的三乙胺和5.0mmol的4-二甲氨基吡啶,0℃~4℃冰浴条件下滴加25.0mmol的乙酰氯,置于40℃条件反应3h,过滤,取有机层,加入饱和食盐水清洗,再加入无水na2so4干燥,减压除去溶剂,柱层析纯化得到淡黄色油状液体3.3g;s2:2-氟-4-肼基苯甲腈的合成:称取10mmol的2,4-二氟苯甲腈加入15ml二氧六环中溶解,再加入15mmol的水合肼,50℃条件下搅拌反应12h,减压旋干,加入饱和食盐水清洗,再加入无水na2so4干燥,减压除去溶剂,加入乙酸乙酯重结晶得到白色固体产物0.89g,熔点136.5-138℃;s3:2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的合成:称取9.9mmol步骤s1得到的化合物5,5-二甲基-2-乙酰基-1,3-环己二酮和9.9mmol步骤s2得到的化合物2-氟-4-肼基苯甲腈加入30ml二氧六环中,再加入15mmol冰乙酸,50℃条件下反应8h,减压去除溶剂,用乙酸乙酯萃取,先加入饱和nahco3清洗,再加入饱和食盐水洗涤,重复清洗2-3次,再加入无水na2so4干燥,浓缩溶剂,加入乙醇重结晶,得到白色固体产物2.7g,熔点149-150℃;s4:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的合成:称取20mmol反式4-羟基环己胺和10mmol步骤s3制得的2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈加至40ml二甲基亚砜中,加入43.5mmol的k2co3,在65℃条件下反应12h,冷却至室温,将反应液加入150ml蒸馏水中,过滤,取滤渣,加入乙醇重结晶,得到固体产物3.7g,熔点192-193℃;s5:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲酰胺的合成:称取2.5mmol步骤s4得到的化合物2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈溶于10ml二氧六环中,再加入0.5mmol的koh和2.6mmol的30%(v/v)的h2o2溶液,在25℃条件下反应2h后,将反应液加入蒸馏水中,过滤,取滤渣,加乙醇重结晶得到产物0.97g,熔点220-222.0℃。合成路线图见图1。实施例3、合成四氢吲唑化合物的方法s1:5,5-二甲基-2-乙酰基1,3-环己二酮的合成:称取20.0mmol的5,5-二甲基-1,3-环己二酮溶于20ml乙酸乙酯中,加入30.0mmol的三乙胺和8.2mmol的4-二甲氨基吡啶,0℃~4℃冰浴条件下滴加30.5mmol的乙酰氯,置于60℃条件反应3h,过滤,取有机层,加入饱和食盐水清洗,再加入无水na2so4干燥,减压除去溶剂,柱层析纯化得到淡黄色油状液体3.5g;s2:2-氟-4-肼基苯甲腈的合成:称取10mmol的2,4-二氟苯甲腈加入15ml乙醇中溶解,再加入12mmol的水合肼,80℃条件下搅拌反应8h,减压旋干,加入饱和食盐水清洗,再加入无水na2so4干燥,减压除去溶剂,加入甲醇重结晶得到白色固体产物0.95g,熔点137-138℃;s3:2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的合成:称取9.9mmol步骤s1得到的化合物5,5-二甲基-2-乙酰基-1,3-环己二酮和9.9mmol步骤s2得到的化合物2-氟-4-肼基苯甲腈加入30ml乙醇中,再加入12.5mmol冰乙酸,80℃条件下反应6h,减压去除溶剂,用乙酸乙酯萃取,先加入饱和nahco3清洗,再加入饱和食盐水洗涤,重复清洗和洗涤2-3次,再加入无水na2so4干燥,浓缩溶剂,加入甲醇重结晶,得到白色固体产物2.67g,熔点150-151℃;s4:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的合成:称取20mmol反式4-羟基环己胺和10mmol步骤s3制得的2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈加至40ml二甲基亚砜中,加入43.5mmol的k2co3,在80℃条件下反应10h,冷却至室温,将反应液加入150ml蒸馏水中,过滤,取滤渣,加入乙醇重结晶,得到固体产物3.8g,熔点191-193℃;s5:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲酰胺的合成:称取2.5mmol步骤s4得到的化合物2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-)苯甲腈溶于10ml乙醇中,再加入0.51mmol的koh和2.6mmol的30%(v/v)的h2o2溶液,在25℃条件下反应2h后,将反应液加入蒸馏水中,过滤,取滤渣,加乙醇重结晶得到产物0.90g,熔点221-222.0℃。合成路线图见图1。试验例一、产率1.实验对象:实施例1-3的合成四氢吲唑化合物的方法。2.根据实施例1-3的合成四氢吲唑化合物的方法,合成目标产物四氢吲唑化合物,计算各中间产物1号:5,5-二甲基-2-乙酰基1,3-环己二酮;2号:2-氟-4-肼基苯甲腈;3号:2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈;4号:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈;5号:2-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲酰胺的合成率(%)。3.实验结果如表1。表1.各实施例中间产物产率(%)产物实施例1实施例2实施例317891952525963374939047795975839588由表1中数据可知,中间产物1:5,5-二甲基-2-乙酰基1,3-环己二酮的最大产率为95%;中间产物2:2-氟-4-肼基苯甲腈的最大产率为63%;中间产物3:2-氟-4-(-3,6,6-三甲基-4-氧代-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的最大产率为93%;中间产物42-(4-羟基环己氨基)-4-(3,6,6-三甲基-4-氧-4,5,6,7-四氢-1h-吲唑-1-基)苯甲腈的最大产率为97%;产物5即四氢吲唑化合物的最大产率为95%。采用该合成路线,可降低最终产物四氢吲唑化合物的合成成本,实现大批量生产以满足市场需求。上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属
技术领域:
中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1